Variant Annotation
Annotate VCF variants with function, clinical significance and pathogenicity.
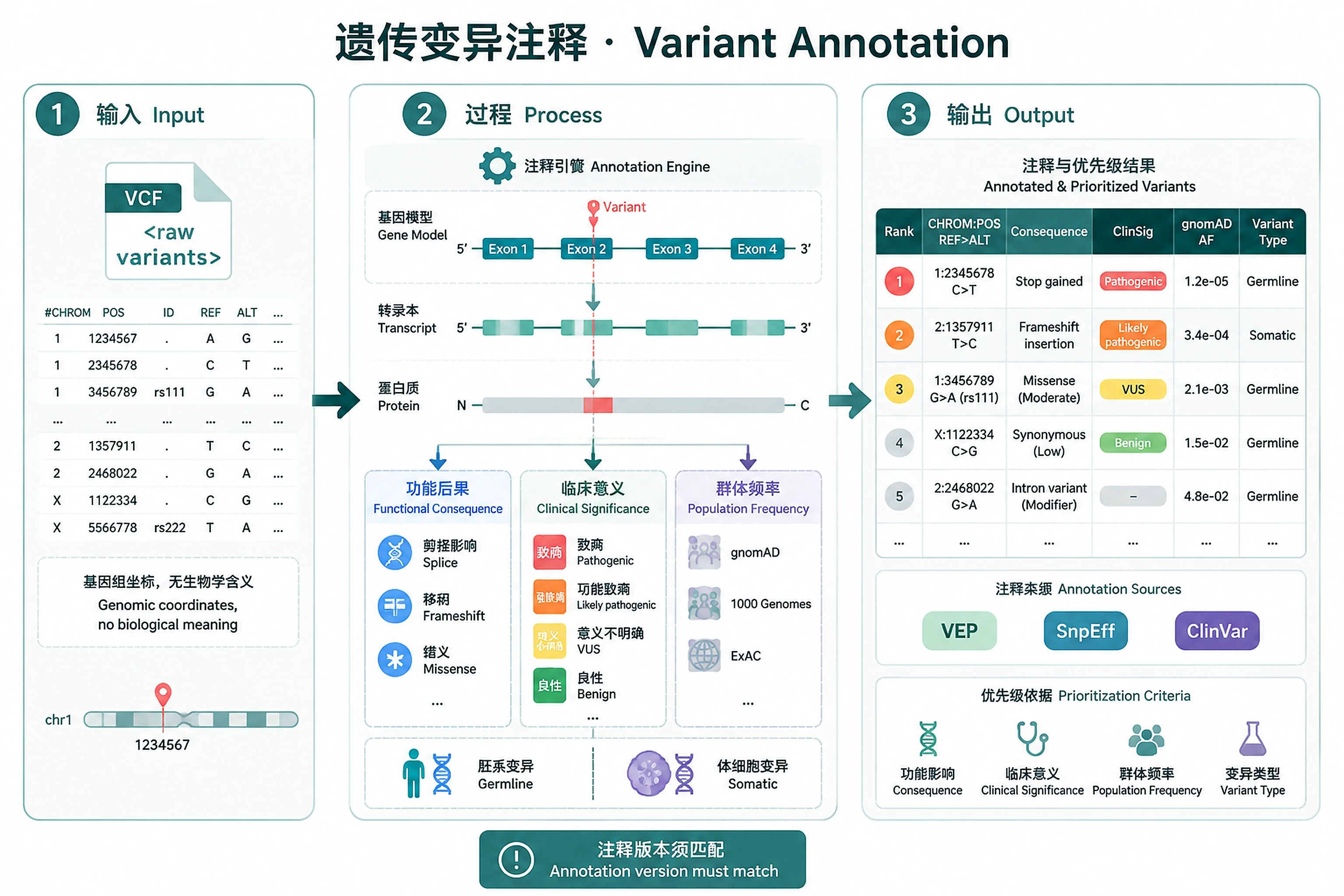
Overview
Problem. A coordinate means nothing until annotated.
Learning goals
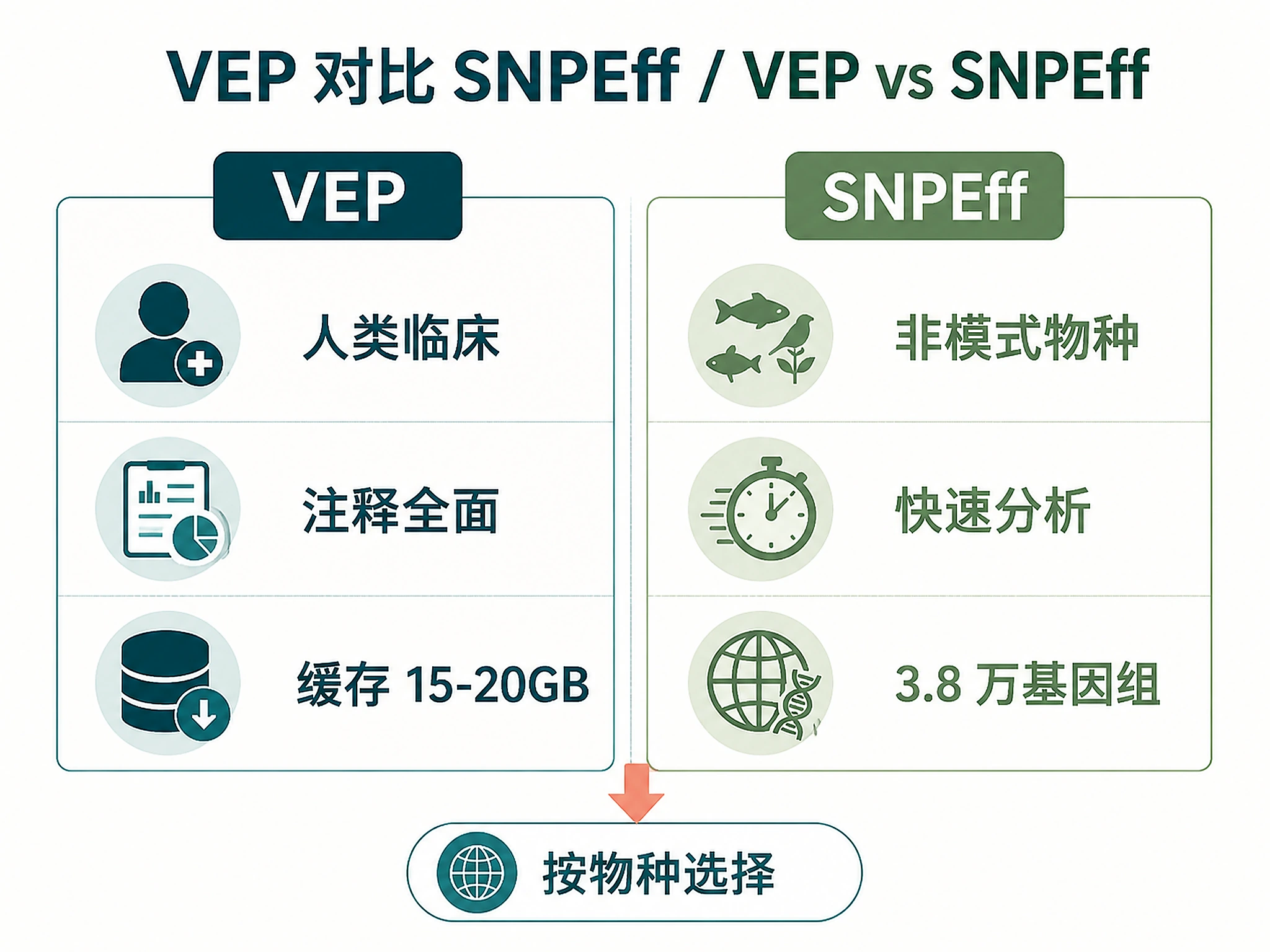
- VEP for human clinical, SnpEff for non-model organisms
- Germline vs somatic interpretation
Figures
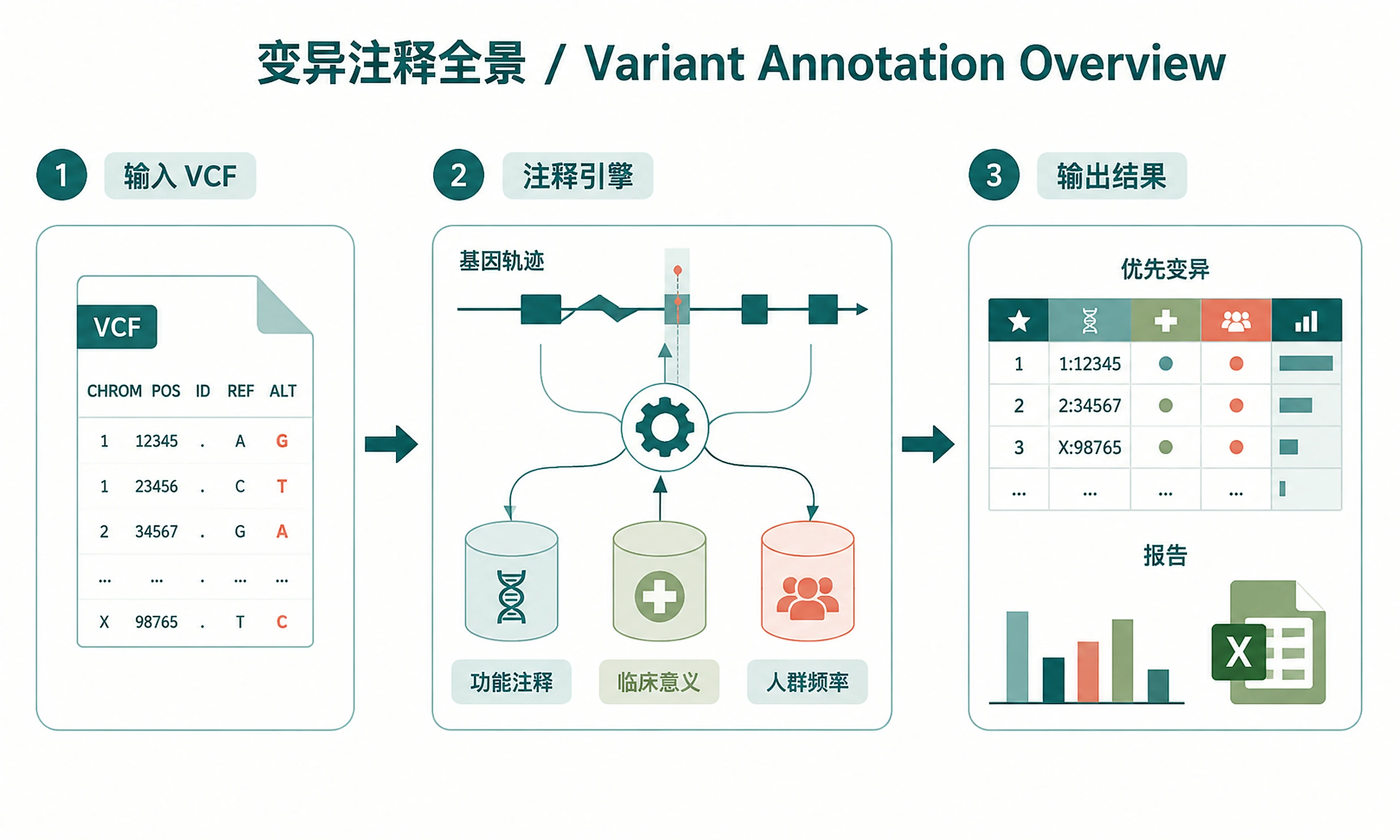
Tutorial
Annotate genomic variants in VCF files with functional effects, clinical significance, and pathogenicity predictions.
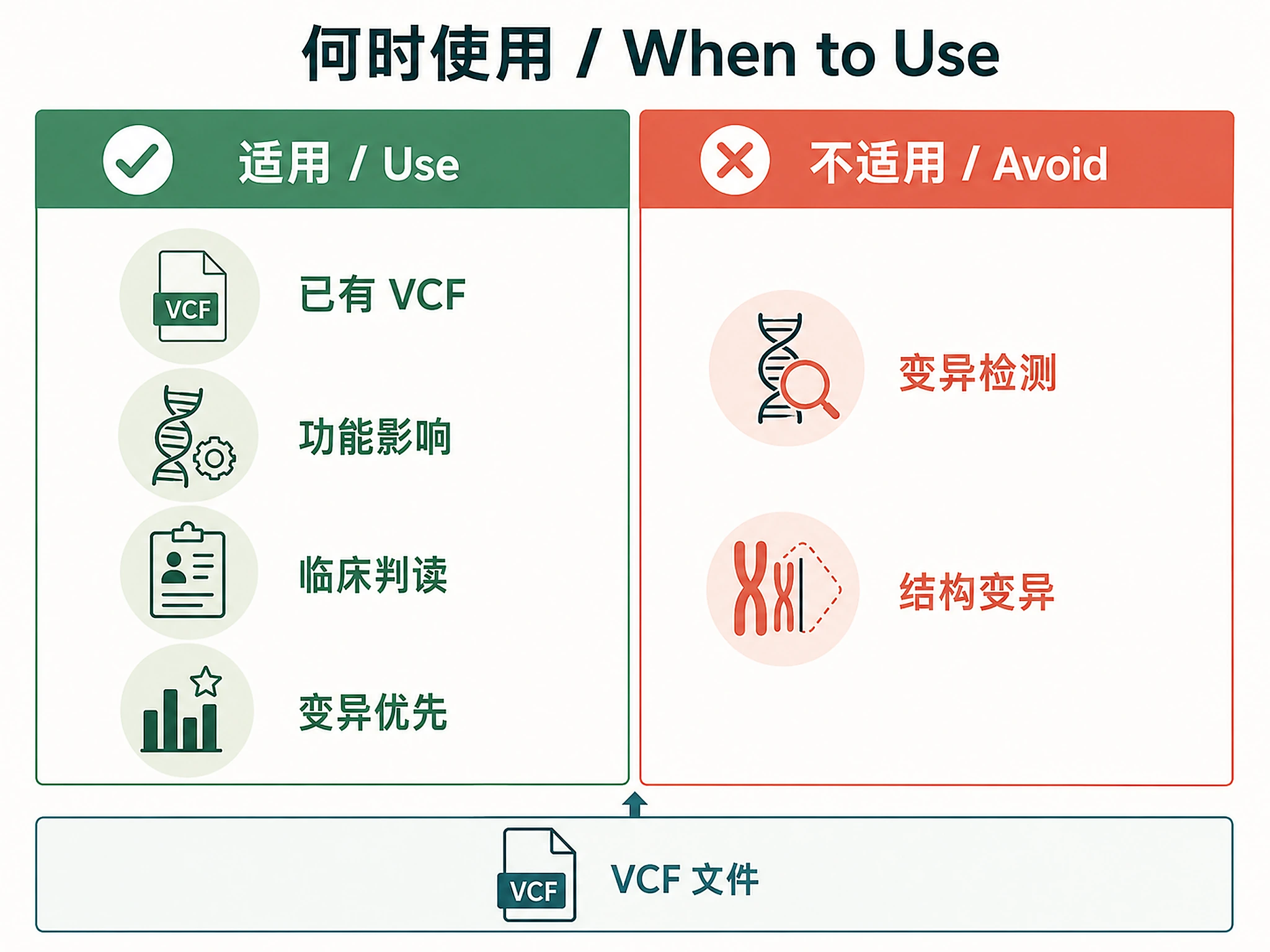
When to Use This Skill
Use this skill when you have:
- ✅ VCF files from variant calling (GATK, bcftools, FreeBayes, etc.)
- ✅ Need functional annotation (gene impact, consequence types)
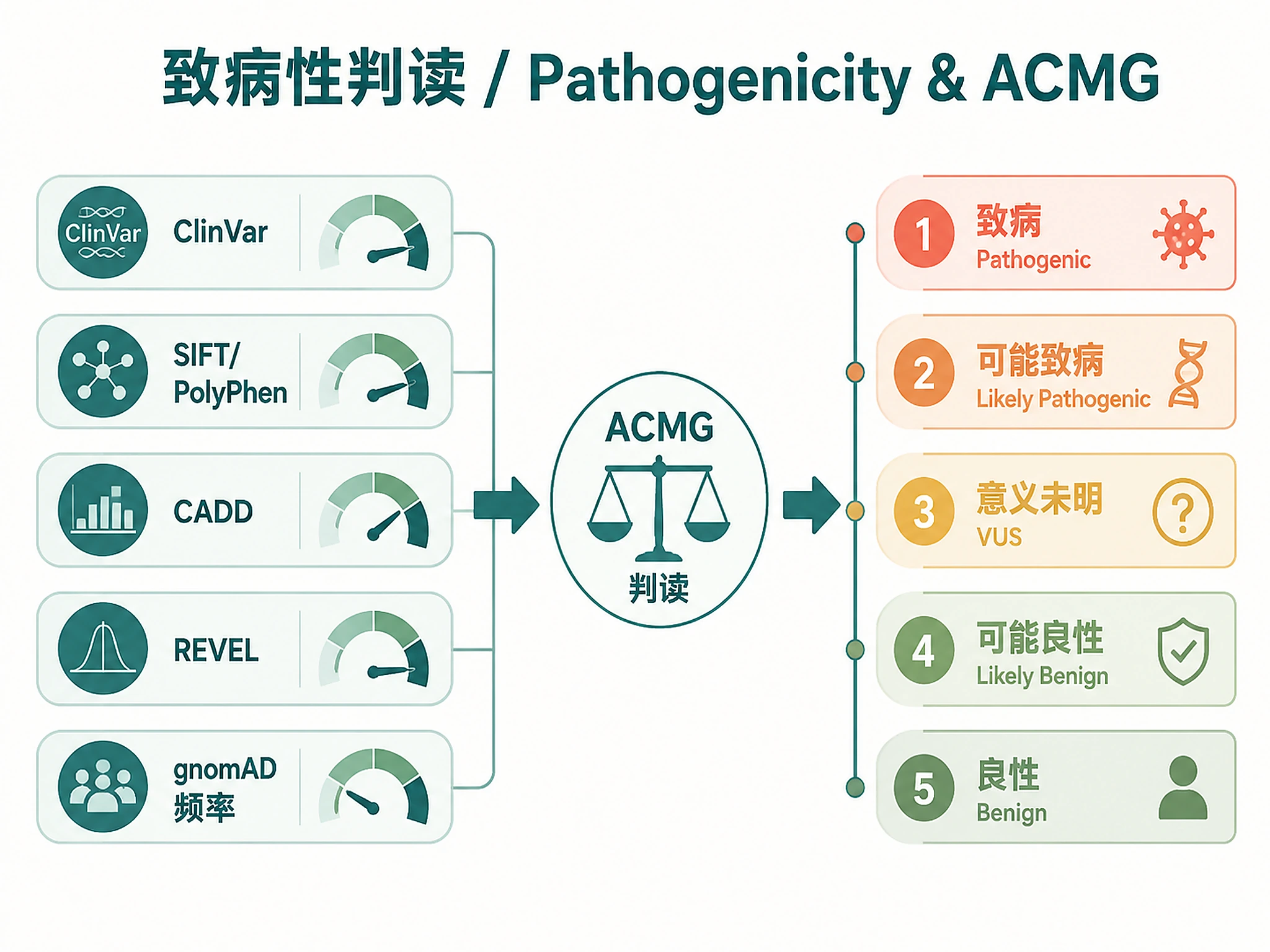
- ✅ Need clinical interpretation (pathogenicity, ClinVar, ACMG classification)
- ✅ Need variant filtering by consequence, frequency, or pathogenicity
- ✅ Working with human, mouse, or 38,000+ other genomes
Use cases:
- Clinical diagnostics (identify pathogenic variants in patient samples)
- Population genetics (annotate with allele frequencies)
- Cancer genomics (somatic variant annotation with COSMIC)
- Research variant prioritization (rank by predicted impact)
- Non-model organism analysis (38,000+ genomes supported)
Don't use for:
- ❌ Variant calling (use GATK/bcftools first) → this skill starts with VCF files
- ❌ Structural variant annotation (limited support, use specialized tools)
Quick Start (Example Data)
Test this skill with example variants:
from scripts.load_example_data import load_clinvar_pathogenic_sample
data = load_clinvar_pathogenic_sample() # Creates test VCF (~10 variants)
print(f"Example VCF created: {data['vcf_path']}")
What you get:
- Dataset: 10 ClinVar pathogenic variants (BRCA1/BRCA2)
- Expected: ~6 HIGH impact, ~8 pathogenic classifications
- Runtime: VEP ~5 min, SNPEff ~2 min
Next steps: Use this VCF in the Standard Workflow below.
Optional validation test (for developers): python assets/eval/simple_test.py to verify installation.
For your own data: See Inputs and Clarification Questions.
Installation
Choose one annotation tool:
| Tool | Best For | Installation | Cache/Database Size |
|---|---|---|---|
| VEP | Human clinical, comprehensive annotations | conda install -c bioconda ensembl-vep |
15-20 GB (one-time) |
| SNPEff | Non-model organisms, quick analysis | conda install -c bioconda snpeff |
2-5 GB per genome |
⚠️ FOR AGENTS: If tools are missing, install them automatically:
# Check and install SNPEff if needed
from scripts.run_snpeff import check_snpeff_installation
is_installed, _, _ = check_snpeff_installation()
if not is_installed:
from scripts.install_tools import install_snpeff
install_snpeff() # Takes ~2-5 min, installs via conda
For VEP:
from scripts.install_tools import install_vep
install_vep() # Takes ~5-10 min, then install cache separately
Python dependencies:
pip install -r requirements.txt
# Or: pip install pandas numpy pysam cyvcf2 plotnine xlsxwriter
Minimum versions:
- Python 3.9+
- VEP 110+ or SNPEff 5.1+
- conda (for tool installation)
VEP cache setup (if using VEP):
vep_install -a c -s homo_sapiens -y GRCh38 # Human, ~15-20 GB, ~30-60 min
SNPEff database setup (if using SNPEff):
snpEff download GRCh38.105 # Human, ~2-5 GB, ~10 min
For detailed installation: See references/installation_guide.md
License: VEP (Apache 2.0), SNPEff (LGPLv3) - both permit commercial use ✅
Inputs
Required:
- VCF file: Variant call format file (bgzipped recommended)
- Must be valid VCF 4.x format
- Coordinate-sorted
- Germline, somatic, or population variants
- SNVs, indels, or mixed (structural variants have limited support)
Genome reference:
- Human: GRCh38/hg38 (recommended) or GRCh37/hg19
- Mouse: GRCm39 or GRCm38
- Other: 38,000+ genomes available in SNPEff
Optional:
- Reference FASTA (for validation)
- BED file (for region-specific annotation)
- Custom gene lists (for filtering)
VCF validation: First workflow step checks format, coordinates, and reference alleles.
Outputs
Analysis objects (Pickle):
analysis_object.pkl- Complete analysis object for downstream use- Load with:
import pickle; obj = pickle.load(open('results/analysis_object.pkl', 'rb')) - Contains: annotated variants DataFrame, gene summaries, tool metadata
- Required for: downstream pathway enrichment, protein structure mapping, literature mining skills
- Size: ~1-10 MB depending on variant count
Annotated variants:
annotated.vep.vcf.gzorannotated.snpeff.vcf.gz- Full VCF with annotations in INFO fieldall_variants.csv- All annotated variants in tabular format
Filtered results:
high_impact_variants.csv- HIGH impact variants onlymoderate_impact_variants.csv- HIGH/MODERATE impact variantsrare_variants.csv- Rare variants (AF < 0.01)filtered_high_impact.vcf.gz- Filtered VCF (maintains VCF format for downstream tools)
Gene summaries:
gene_summary.csv- Variants aggregated per gene (useful for pathway enrichment)
Visualizations (PNG + SVG, 300 DPI):
consequence_distribution.png/.svg- Variant types (missense, stop_gained, etc.)impact_by_chromosome.png/.svg- Impact severity distribution across chromosomespathogenicity_scores.png/.svg- CADD, REVEL, SIFT score distributionsallele_frequency.png/.svg- Population frequency distributiongene_burden.png/.svg- Top genes by variant burden
Reports:
summary_report.xlsx- Excel report with multiple sheets (summary statistics, top consequences, top genes, variants)
Clarification Questions
⚠️ CRITICAL: Always ask question #1 first to check if user has provided VCF files.
Before starting, gather:
-
Input Files (ASK THIS FIRST):
- Do you have specific VCF file(s) to annotate?
- If uploaded: Is this the VCF you'd like to annotate?
- Expected format: VCF/VCF.GZ (variant call format)
- Or use example data for testing?
- Use
load_clinvar_pathogenic_sample()(~10 variants, known pathogenic)
-
Organism and Genome Build:
- What organism? (human, mouse, zebrafish, etc.)
- Which genome assembly? (GRCh38/hg38 recommended for human)
- ⚠️ Must match VCF reference - check VCF header:
##reference=
-
Primary Use Case (determines tool selection):
- Clinical/medical genetics → VEP (comprehensive clinical databases)
- Population genetics → Either tool works
- Non-model organism → SNPEff (38,000+ genomes)
- Quick analysis → SNPEff (faster setup)
- See references/tool_selection_guide.md for detailed comparison
-
Annotation Priorities (select all that apply):
- Clinical significance (ClinVar, OMIM)
- Pathogenicity predictions (SIFT, PolyPhen, CADD, REVEL)
- Population frequencies (gnomAD, 1000 Genomes)
- Regulatory impacts (ENCODE)
- Basic consequences only (faster annotation)
-
Computational Resources:
- High-performance (32+ GB RAM) → VEP with full cache
- Standard (16-32 GB) → VEP or SNPEff
- Limited (<16 GB) → SNPEff recommended
-
Output Requirements:
- Variant prioritization? → Apply filtering + ACMG classification
- Gene-level analysis? → Generate gene summaries
- Clinical reporting? → Excel export with multiple sheets
- Statistical analysis? → CSV format for downstream tools
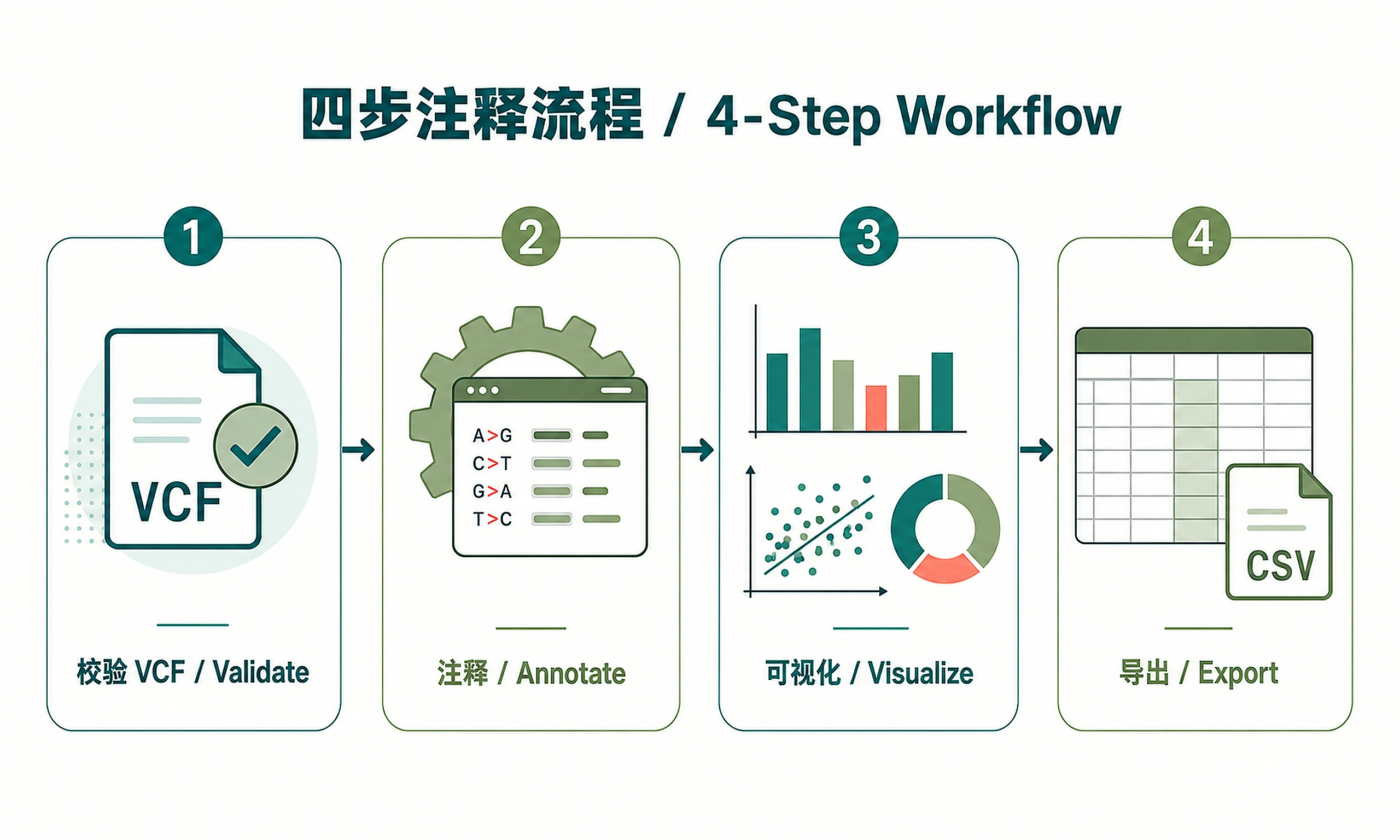
Standard Workflow
🚨 MANDATORY: USE SCRIPTS EXACTLY AS SHOWN - DO NOT WRITE INLINE CODE 🚨
CRITICAL: Use relative paths (scripts/). DO NOT construct absolute paths like /mnt/knowhow/ or /workspace/.
This is a 4-step workflow. Scripts handle all complexity automatically.
Step 1 - Load and validate data:
# Load example data (or use your own VCF file)
from scripts.load_example_data import load_clinvar_pathogenic_sample
data = load_clinvar_pathogenic_sample()
input_vcf = data['vcf_path']
# Validate VCF format
from scripts.validate_vcf import validate_vcf, vcf_summary_stats
results = validate_vcf(input_vcf)
if not results['is_valid']:
print(f"Errors: {results['errors']}")
stats = vcf_summary_stats(input_vcf)
print(f"Total: {stats['total_variants']}, SNVs: {stats['snvs']}, Indels: {stats['indels']}")
✅ VERIFICATION: You MUST see: "✓ Data loaded successfully"
DO NOT write inline validation code. Use the scripts exactly as shown.
Step 2 - Run annotation:
# Select and run annotation tool
from scripts.select_tool import select_annotation_tool
tool = select_annotation_tool(organism='human', use_case='clinical', resources='standard')
# Check if tool is installed, install if needed
if tool == 'vep':
from scripts.run_vep import check_vep_installation
is_installed, _, _ = check_vep_installation()
if not is_installed:
print("VEP not found. Installing via conda...")
from scripts.install_tools import install_vep
install_vep()
# Note: After VEP install, you must install cache separately
print("\n⚠️ IMPORTANT: Now install VEP cache:")
print(" vep_install -a c -s homo_sapiens -y GRCh38")
elif tool == 'snpeff':
from scripts.run_snpeff import check_snpeff_installation
is_installed, _, _ = check_snpeff_installation()
if not is_installed:
print("SNPEff not found. Installing via conda...")
from scripts.install_tools import install_snpeff
install_snpeff()
# VEP annotation
if tool == 'vep':
from scripts.run_vep import run_vep
annotated_vcf = run_vep(input_vcf, "annotated.vep.vcf.gz", genome="GRCh38", everything=True, fork=4)
from scripts.parse_vep_output import parse_vep_vcf
df = parse_vep_vcf("annotated.vep.vcf.gz")
# SNPEff annotation
elif tool == 'snpeff':
from scripts.run_snpeff import run_snpeff
annotated_vcf = run_snpeff(input_vcf, "annotated.snpeff.vcf.gz", genome="GRCh38.105", canon=True, threads=4)
from scripts.parse_snpeff_output import parse_snpeff_vcf
df = parse_snpeff_vcf("annotated.snpeff.vcf.gz")
✅ VERIFICATION: You MUST see: "✓ Annotation completed successfully!"
DO NOT write inline annotation code. Use run_vep() or run_snpeff() as shown.
DO NOT fall back to VEP API mode
API mode is 10-100x slower and less full-featured. Always install the tool if missing.
Step 3 - Generate visualizations:
from scripts.filter_variants import filter_by_consequence, filter_by_frequency
from scripts.annotate_genes import annotate_gene_summary
from scripts.plot_variant_distribution import (
plot_consequence_distribution,
plot_impact_by_chromosome,
plot_pathogenicity_scores,
plot_allele_frequency,
plot_gene_burden
)
# Filter variants for analysis
high_impact = filter_by_consequence(df, consequence_levels=['HIGH', 'MODERATE'])
rare = filter_by_frequency(df, max_af=0.01)
# Generate gene-level summaries
gene_df = annotate_gene_summary(df)
# Create visualizations (PNG + SVG)
output_dir = "results"
plot_consequence_distribution(df, f"{output_dir}/consequence_distribution.png")
plot_impact_by_chromosome(df, f"{output_dir}/impact_by_chromosome.png")
plot_pathogenicity_scores(df, f"{output_dir}/pathogenicity_scores.png")
plot_allele_frequency(df, f"{output_dir}/allele_frequency.png")
if gene_df is not None:
plot_gene_burden(gene_df, output_file=f"{output_dir}/gene_burden.png")
✅ VERIFICATION: You MUST see: "Saved: [filename].webp" and "Saved: [filename].svg" for each plot
DO NOT write inline plotting code. Use the plotting functions as shown.
Step 4 - Export results:
from scripts.export_results import export_all
# Export all results in all formats
export_all(
df=df,
gene_df=gene_df,
original_vcf=input_vcf,
output_dir="results",
tool_name=tool
)
✅ VERIFICATION: You MUST see: "=== Export Complete ==="
DO NOT write custom export code. Use export_all() as shown.
⚠️ CRITICAL - DO NOT:
- ❌ Write inline validation/annotation/plotting/export code → STOP: Use the scripts shown above
- ❌ Fall back to VEP API mode when tools are missing → STOP: Use
install_tools.pyto install via conda - ❌ Use absolute paths like
/mnt/knowhow/or/workspace/→ use relative pathsscripts/ - ❌ Skip validation step → catches format errors early
- ❌ Mix genome builds → VCF and annotation database must match (check VCF header)
- ❌ Write custom export code → STOP: Use
export_all()
If tools are missing: Use install_tools.py to install via conda (~2-10 min). DO NOT use VEP API mode (10-100x slower, less full-featured).
If scripts fail: Install missing dependencies, re-run. Only modify scripts if genuinely broken.
Common Issues
| Issue | Possible Cause | Solution | Details |
|---|---|---|---|
| VEP/SNPEff fails to start | Tool not installed or not in PATH | Use install_tools.py: from scripts.install_tools import install_snpeff; install_snpeff() |
Takes 2-5 min. DO NOT fall back to VEP API mode (10-100x slower). Verify with which vep or which snpEff |
| "Cannot find genome" | Database not downloaded | Run install_vep_cache() or download_snpeff_database() |
See installation_guide.md |
| All variants annotated as intergenic | Genome build mismatch (hg19 vs hg38) | Check VCF header ##reference=, use matching genome build |
Use bcftools annotate --rename-chrs to convert |
| No pathogenicity scores | Plugins/databases not installed | Install VEP plugins or SnpSift databases | See vep_best_practices.md |
| Memory error during annotation | Insufficient RAM or large VCF | Reduce buffer size (--buffer_size 1000) or split VCF by chromosome |
Use bcftools view -r chr1 to split |
| "Invalid VCF format" | VCF format errors (tabs, header, etc.) | Run validation step first, check tabs and header completeness | Use bcftools view -h to inspect header |
| Chromosome name mismatch | VCF uses "chr1" but database uses "1" | Convert with bcftools annotate --rename-chrs |
Create mapping file: chr1 1\nchr2 2\n... |
| Very slow annotation | Not using cache/database (API mode) | Ensure VEP cache or SNPEff database installed locally | API mode is 10-100x slower |
For complete troubleshooting: See references/troubleshooting_guide.md
QC checkpoints:
- VCF validation: Ti/Tv ratio 2.0-2.1 (genome) or 2.8-3.0 (exome)
- Annotation completeness: 100% variants annotated
- Expected HIGH impact: 10-50 per exome (healthy individual)
For detailed QC thresholds: See references/qc_guidelines.md
Suggested Next Steps
After annotating variants:
-
Variant prioritization → Filter and rank by pathogenicity
- Use filtering: AF < 0.01, HIGH/MODERATE impact, CADD > 20
- Apply ACMG classification for clinical reporting
-
Gene-level analysis → Aggregate variants per gene
- Identify genes with high variant burden
- Prepare for pathway enrichment analysis
-
Clinical reporting → Export prioritized variants
- Excel format with multiple sheets (variants, genes, summary)
- Include ACMG classifications and ClinVar annotations
-
Statistical analysis → Burden testing, association studies
- Compare variant frequencies between cases and controls
- Gene-based collapsing analysis
Related analyses: Pathway enrichment from gene lists, protein structure mapping, literature mining
Related Skills
Upstream (run these first):
- Variant calling workflows (GATK HaplotypeCaller, bcftools call, FreeBayes)
- Variant quality score recalibration (VQSR)
Downstream (run these after):
- Pathway enrichment from gene lists
- Protein structure visualization and impact prediction
- Clinical report generation
Complementary:
- Copy number variant (CNV) annotation
- Structural variant annotation (specialized tools)
- Pharmacogenomics annotation (PharmGKB)
References
Primary Citations
Ensembl VEP:
- McLaren W, et al. (2016) The Ensembl Variant Effect Predictor. Genome Biology 17:122. doi:10.1186/s13059-016-0974-4
SNPEff:
- Cingolani P, et al. (2012) A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff. Fly 6(2):80-92. doi:10.4161/fly.19695
ACMG Guidelines:
- Richards S, et al. (2015) Standards and guidelines for the interpretation of sequence variants. Genetics in Medicine 17(5):405-424. PubMed:25741868
Documentation
Tool Selection & Configuration:
- Tool Selection Guide - VEP vs SNPEff decision matrix, resource requirements
- VEP Best Practices - VEP configuration, plugins, optimization
- SNPEff Best Practices - SNPEff parameters, databases, statistics
Analysis & Interpretation:
- QC Guidelines - Quality control metrics, expected ranges, red flags
- Consequence Terms - Sequence Ontology terms reference
- Pathogenicity Interpretation - ACMG/AMP guidelines, classification rules
- Filtering Strategies - Clinical vs research filtering approaches
Setup & Troubleshooting:
- Installation Guide - Detailed VEP/SNPEff installation instructions
- Troubleshooting Guide - Common issues, diagnosis steps, solutions
Code preview
scripts/annotate_genes.py
"""
Gene-Level Annotation Module
This module provides functions for generating gene-level summaries from variant data.
"""
import sys
from collections import defaultdict
try:
import pandas as pd
import numpy as np
except ImportError as e:
print(f"Error: Missing required package: {e}")
print("Install with: pip install pandas numpy")
sys.exit(1)
def create_gene_summary(
df,
group_by='SYMBOL',
count_variants=True,
count_by_consequence=True,
count_by_impact=True,
include_most_severe=True,
aggregate_scores=None,
aggregation='max'
):
"""
Create gene-level summary from variant data.
Parameters
----------
df : pd.DataFrame
DataFrame with variant annotations
group_by : str
Column to group by (default: 'SYMBOL' for gene symbol)
count_variants : bool
Count total variants per gene
count_by_consequence : bool
Count variants by consequence type
count_by_impact : bool
Count variants by impact level
include_most_severe : bool
Include most severe variant per gene
aggregate_scores : list, optional
List of score columns to aggregate (e.g., ['CADD_PHRED', 'REVEL'])
aggregation : str
Aggregation method: 'max', 'mean', 'median' (default: 'max')
Returns
-------
pd.DataFrame
Gene-level summary DataFrame
"""
if group_by not in df.columns:
raise ValueError(f"Column '{group_by}' not found in DataFrame")
# Remove missing gene names
df_genes = df[df[group_by].notna()].copy()
# Initialize summary dict
summary_data = defaultdict(dict)
# Group by gene
for gene, gene_df in df_genes.groupby(group_by):
gene_summary = {}
# Total variant count
if count_variants:
gene_summary['N_Variants'] = len(gene_df)
# Count by impact
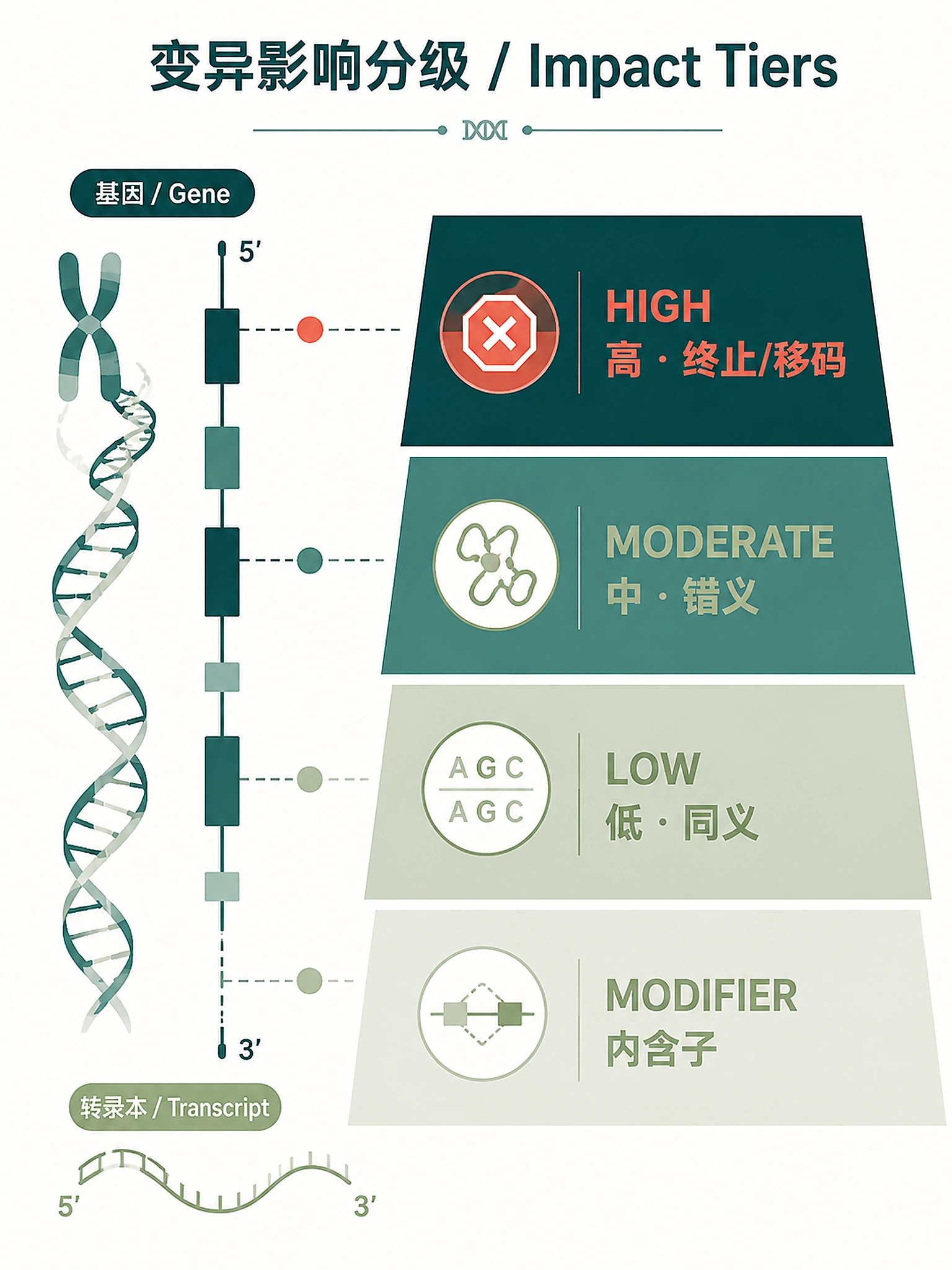
if count_by_impact and 'IMPACT' in gene_df.columns:
for impact in ['HIGH', 'MODERATE', 'LOW', 'MODIFIER']:
count = len(gene_df[gene_df['IMPACT'] == impact])
gene_summary[f'N_{impact}_Impact'] = count
# Count by consequence
if count_by_consequence and 'Consequence' in gene_df.columns:scripts/export_results.py
"""
Export Results Module
This module provides functions for exporting annotated variants in multiple formats.
"""
import sys
from pathlib import Path
try:
import pandas as pd
import xlsxwriter
from openpyxl import load_workbook
from openpyxl.styles import Font, PatternFill, Alignment
except ImportError as e:
print(f"Error: Missing required package: {e}")
print("Install with: pip install pandas xlsxwriter openpyxl")
sys.exit(1)
def export_to_excel(output_file, sheets, format_columns=True, freeze_panes=(1, 2)):
"""
Export data to Excel with multiple sheets and formatting.
Parameters
----------
output_file : str
Output Excel file path
sheets : dict
Dictionary mapping sheet names to DataFrames
format_columns : bool
Apply conditional formatting (default: True)
freeze_panes : tuple
Row and column to freeze (default: (1, 2) for header + 2 columns)
"""
# Create Excel writer
with pd.ExcelWriter(output_file, engine='xlsxwriter') as writer:
workbook = writer.book
# Define formats
header_format = workbook.add_format({
'bold': True,
'bg_color': '#D9E1F2',
'border': 1,
'align': 'center',
'valign': 'vcenter'
})
high_impact_format = workbook.add_format({'bg_color': '#FFC7CE', 'font_color': '#9C0006'})
moderate_impact_format = workbook.add_format({'bg_color': '#FFEB9C', 'font_color': '#9C6500'})
pathogenic_format = workbook.add_format({'bg_color': '#FFC7CE', 'font_color': '#9C0006'})
# Write each sheet
for sheet_name, df in sheets.items():
df.to_excel(writer, sheet_name=sheet_name, index=False, startrow=0)
worksheet = writer.sheets[sheet_name]
# Format header
for col_num, column_name in enumerate(df.columns):
worksheet.write(0, col_num, column_name, header_format)
# Auto-fit columns
for col_num, column_name in enumerate(df.columns):
column_data = df[column_name].astype(str)
max_length = max(
column_data.apply(len).max(),
len(column_name)
) + 2
worksheet.set_column(col_num, col_num, min(max_length, 50))
# Freeze panes
if freeze_panes:
worksheet.freeze_panes(freeze_panes[0], freeze_panes[1])
# Conditional formatting
if format_columns and 'IMPACT' in df.columns:
impact_col = df.columns.get_loc('IMPACT')
worksheet.conditional_format(
1, impact_col, len(df), impact_col,
{scripts/filter_variants.py
"""
Variant Filtering Module
This module provides functions for filtering annotated variants by consequence,
frequency, quality, and pathogenicity.
"""
import sys
try:
import pandas as pd
import numpy as np
except ImportError as e:
print(f"Error: Missing required package: {e}")
print("Install with: pip install pandas numpy")
sys.exit(1)
# Consequence severity definitions (Sequence Ontology)
HIGH_IMPACT_CONSEQUENCES = {
'transcript_ablation',
'splice_acceptor_variant',
'splice_donor_variant',
'stop_gained',
'frameshift_variant',
'stop_lost',
'start_lost',
'transcript_amplification'
}
MODERATE_IMPACT_CONSEQUENCES = {
'inframe_insertion',
'inframe_deletion',
'missense_variant',
'protein_altering_variant'
}
LOW_IMPACT_CONSEQUENCES = {
'splice_region_variant',
'incomplete_terminal_codon_variant',
'start_retained_variant',
'stop_retained_variant',
'synonymous_variant'
}
def filter_by_consequence(df, impact=None, consequence_types=None):
"""
Filter variants by consequence severity or specific consequence types.
Parameters
----------
df : pd.DataFrame
DataFrame with variant annotations
impact : list, optional
List of impact levels to keep: ['HIGH', 'MODERATE', 'LOW', 'MODIFIER']
consequence_types : list, optional
List of specific consequence types to keep
Returns
-------
pd.DataFrame
Filtered DataFrame
"""
filtered = df.copy()
# Filter by IMPACT
if impact:
if 'IMPACT' not in df.columns:
print("Warning: IMPACT column not found")
return filtered
filtered = filtered[filtered['IMPACT'].isin(impact)]
# Filter by specific consequence types
if consequence_types:
if 'Consequence' not in df.columns:
print("Warning: Consequence column not found")
return filtered