Cell–Cell Communication
Infer ligand–receptor communication networks with CellChat v2.
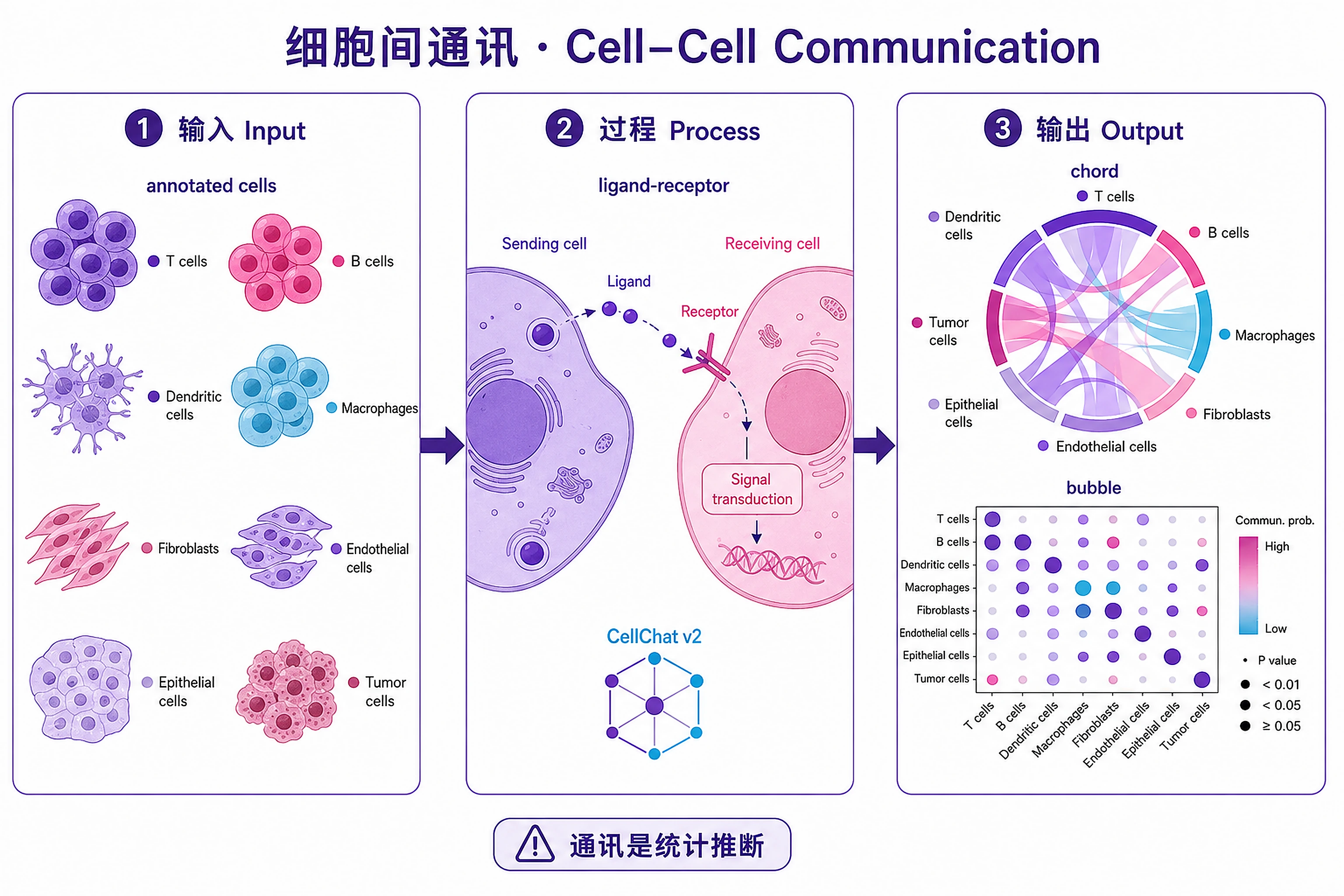
Overview
Problem. Which pathways let cell groups talk to each other.
Learning goals
- Communication is statistical, not observed
- Learn to read chord and bubble plots
Figures
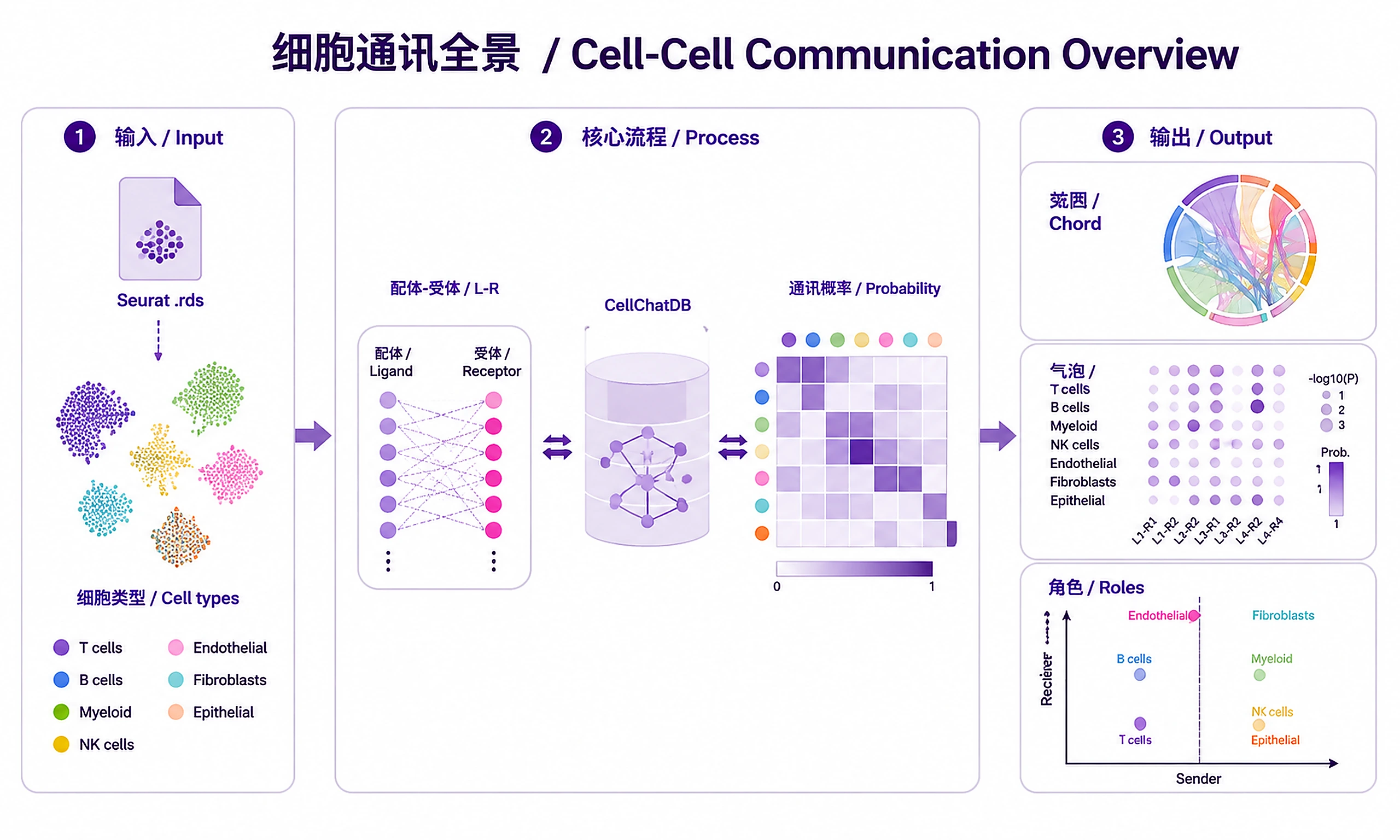
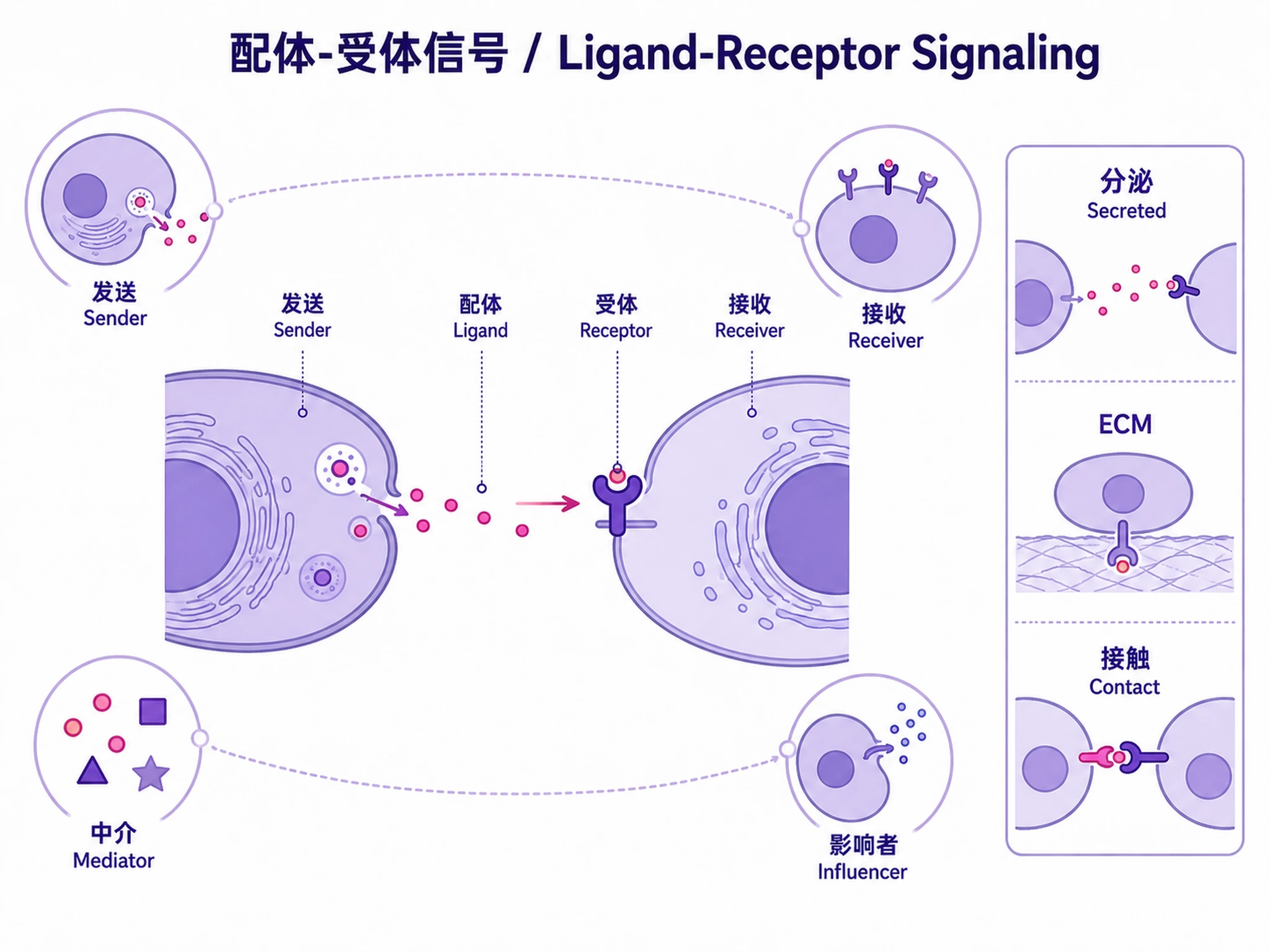
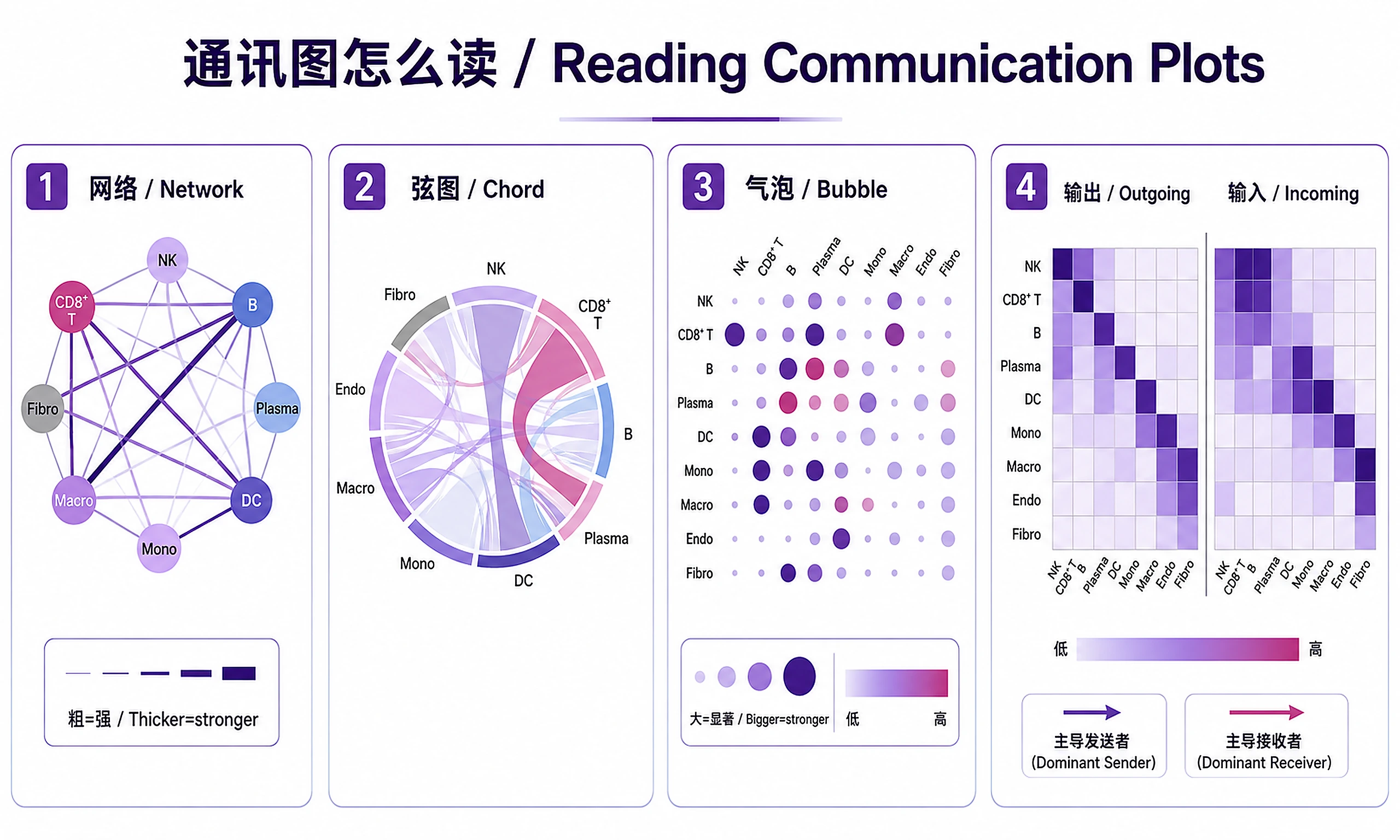
Tutorial
When to Use This Skill
✅ Use when:
- You have an annotated scRNA-seq dataset (Seurat object with cell type labels)
- You want to identify ligand-receptor interactions between cell types
- You want to visualize communication networks (chord diagrams, bubble plots)
- You want to find dominant sender/receiver cell populations
- Chains from
scrnaseq-seurat-core-analysisoutput (seurat_processed.rds)
❌ Don't use when:
- Data is not annotated (run
scrnaseq-seurat-core-analysisfirst) - You need spatial cell-cell communication (CellChat v2 supports this but requires spatial coordinates)
- You want gene regulatory networks (use
grn-pyscenicinstead) - You have bulk RNA-seq data
Installation
| Package | Version | License | Commercial Use | Installation |
|---|---|---|---|---|
| CellChat | ≥2.0.0 | GPL-3 | ✅ Permitted | devtools::install_github("jinworks/CellChat") |
| Seurat | ≥5.0.0 | MIT | ✅ Permitted | install.packages('Seurat') |
| SeuratData | ≥0.2.1 | GPL-3 | ✅ Permitted | devtools::install_github('satijalab/seurat-data') |
| NMF | ≥0.23.0 | GPL-2+ | ✅ Permitted | install.packages('NMF') |
| circlize | ≥0.4.12 | MIT | ✅ Permitted | install.packages('circlize') |
| ComplexHeatmap | ≥2.12.0 | MIT | ✅ Permitted | BiocManager::install('ComplexHeatmap') |
| ggprism | ≥1.0.3 | GPL-3 | ✅ Permitted | install.packages('ggprism') |
| presto | ≥1.0.0 | GPL-3 | ✅ Permitted | remotes::install_github('immunogenomics/presto') |
| ggalluvial | ≥0.12.0 | GPL-2 | ✅ Permitted | install.packages('ggalluvial') |
| rmarkdown | ≥2.20 | GPL-3 | ✅ Permitted | install.packages('rmarkdown') |
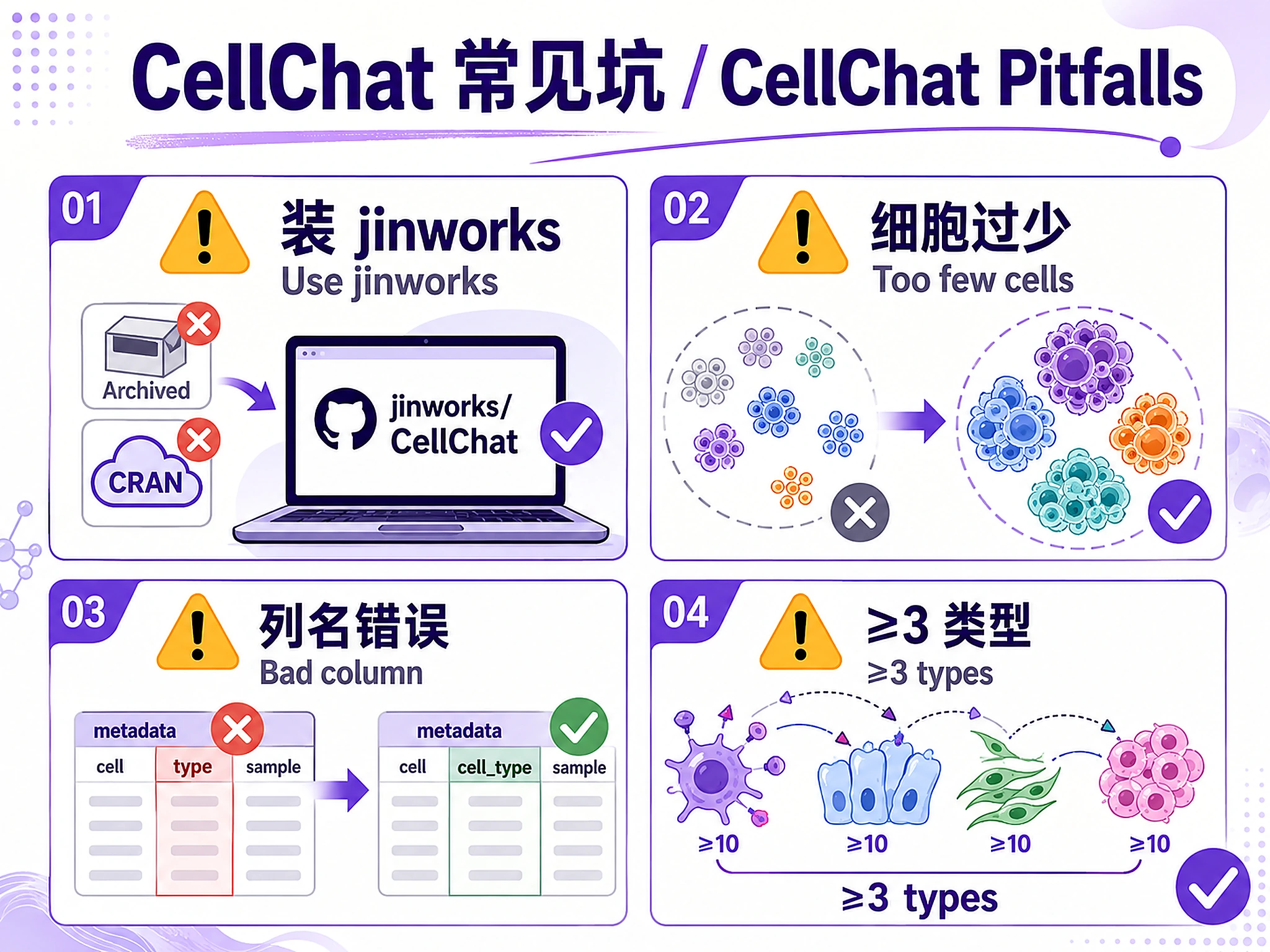
⚠️ CellChat must be installed from GitHub (not CRAN). Use the jinworks repository (active), not sqjin (archived).
Inputs
Required:
- Seurat object (.rds) with:
- Normalized expression data (
@assays$RNA@data) - Cell type annotations in metadata (e.g.,
celltypecolumn) - Minimum 3 cell types, ≥10 cells per type recommended
Accepted sources:
seurat_processed.rdsfromscrnaseq-seurat-core-analysis(chains directly)- Any annotated Seurat v5 object
- Example PBMC data (auto-loaded if no file provided)
Outputs
CSV tables:
significant_interactions.csv— All significant L-R pairs with source, target, pathway, probabilitypathway_summary.csv— Pathway-level communication summaryinteraction_count_matrix.csv— Cell type × cell type interaction countsinteraction_strength_matrix.csv— Cell type × cell type communication weightssignaling_roles.csv— Centrality scores (sender, receiver, mediator, influencer per pathway)top_interactions.csv— Top 20 interactions ranked by probability
Visualizations (PNG + SVG):
interaction_count_network— Circle plot of interaction countsinteraction_strength_network— Circle plot of communication strengthchord_aggregated— Chord diagram of the full communication networkbubble_ligand_receptor— Bubble plot of L-R pairs by cell type pairssignaling_outgoing_heatmap— Outgoing signaling patterns by cell typesignaling_incoming_heatmap— Incoming signaling patterns by cell typesignaling_role_scatter— Dominant senders vs receivers scatter
Analysis objects (RDS):
cellchat_object.rds— Complete CellChat object for downstream use- Load with:
cellchat <- readRDS('cellchat_object.rds') - Required for: multi-condition comparison, pathway-specific deep dives
Reports:
analysis_report.md— Markdown report (always generated)analysis_report.pdf— PDF report (requires rmarkdown + LaTeX)
Clarification Questions
🚨 ALWAYS ask Question 1 FIRST. Do not proceed before the user answers.
1. Input Files (ASK THIS FIRST):
- Do you have an annotated Seurat object (.rds) from scRNA-seq analysis?
- If yes: provide the path to the
.rdsfile - Expected: Seurat v5 object with cell type labels in metadata
- Or use example data? — PBMC 3k dataset (human immune cells, 2,638 cells, 8 cell types)
- Uses
source("scripts/load_data.R"); seurat_obj <- load_example_pbmc()
🚨 IF EXAMPLE DATA SELECTED: Parameters are pre-defined. Skip to Question 4 (or proceed directly to Step 1). Do NOT ask questions 2-3.
2. Species (own data only):
- a) Human (CellChatDB.human) — default
- b) Mouse (CellChatDB.mouse)
3. Cell Type Column (own data only):
- Which metadata column contains cell type annotations?
- Common:
celltype,singler_labels,cell_type,predicted.celltype.l2 - Check with:
colnames(seurat_obj@meta.data)
4. Analysis Scope (structured — works for demo and own data):
- a) All signaling types (Secreted + ECM-Receptor + Cell-Cell Contact) — ✅ recommended
- b) Secreted signaling only
- c) Cell-Cell Contact only
Standard Workflow
🚨 MANDATORY: USE SCRIPTS EXACTLY AS SHOWN — DO NOT WRITE INLINE CODE 🚨
Step 1 — Load data:
source("scripts/load_data.R")
seurat_obj <- load_cellchat_data() # example PBMC data
# OR: seurat_obj <- load_cellchat_data("path/to/seurat_processed.rds")
Step 2 — Run CellChat analysis:
source("scripts/run_cellchat.R")
cellchat <- run_cellchat_analysis(seurat_obj, species = "human", group.by = "celltype")
DO NOT write inline CellChat code. Just source the script and call the function.
Step 3 — Generate visualizations:
source("scripts/cellchat_plots.R")
generate_all_plots(cellchat, output_dir = "results")
DO NOT write inline plotting code. Just use the script.
Step 4 — Export results:
source("scripts/export_results.R")
export_all(cellchat, seurat_obj = seurat_obj, output_dir = "results")
DO NOT write custom export code. Use export_all().
✅ VERIFICATION — You should see:
- After Step 1:
"✓ Data loaded successfully! [N] cells, [M] cell types" - After Step 2:
"✓ CellChat analysis completed! [N] significant interactions across [M] pathways" - After Step 3:
"✓ All plots generated successfully! [6] visualizations saved" - After Step 4:
"=== Export Complete ==="
❌ IF YOU DON'T SEE THESE: You wrote inline code. Stop and use source().
⚠️ CRITICAL — DO NOT:
- ❌ Write inline CellChat code → STOP: Use
source("scripts/run_cellchat.R") - ❌ Write inline plotting code → STOP: Use
generate_all_plots() - ❌ Write custom export code → STOP: Use
export_all() - ❌ Try to install system-level dependencies → CellChat handles its own deps
⚠️ IF SCRIPTS FAIL — Script Failure Hierarchy:
- Fix and Retry (90%) — Install missing package, re-run script
- Modify Script (5%) — Edit the script file itself, document changes
- Use as Reference (4%) — Read script, adapt approach, cite source
- Write from Scratch (1%) — Only if genuinely impossible, explain why
NEVER skip directly to writing inline code without trying the script first.
Common Issues
| Issue | Cause | Solution |
|---|---|---|
| CellChat not found | Not installed from GitHub | devtools::install_github("jinworks/CellChat") — must use jinworks repo (not sqjin) |
| "presto" required for Wilcoxon test | Missing presto package | remotes::install_github('immunogenomics/presto') — script falls back to standard test if unavailable |
| No significant interactions | Too few cells per type or stringent filtering | Lower min.cells parameter or merge rare cell types |
| Memory error on large datasets | >50k cells uses substantial RAM | Subsample or increase memory; see references/cellchat-guide.md |
| Chord diagram error | Missing circlize package | install.packages('circlize') |
| SVG export error "svglite required" | Missing optional dependency | Use generate_all_plots() — it handles fallback automatically. DO NOT try to install svglite manually. |
| svglite dependency conflict | System library version mismatch | Normal — generate_all_plots() falls back to base R svg() device automatically. Both PNG and SVG will be created. |
| "group.by not found" | Wrong column name for cell types | Check: colnames(seurat_obj@meta.data) |
| Seurat v5 slot error ("no slot of name images") | Old Seurat object from v3/v4 | Script handles this — UpdateSeuratObject() is called automatically |
| NMF not available | NMF package not installed | install.packages('NMF') |
| PDF report skipped | No LaTeX installation | install.packages('tinytex'); tinytex::install_tinytex() — markdown report still available |
Suggested Next Steps
After cell-cell communication analysis, consider:
- Multi-condition comparison — Compare communication between disease vs healthy, treated vs untreated
- See references/cellchat-guide.md for
mergeCellChat()workflow - Pathway deep dive — Examine specific pathways (e.g., TNF, MHC-II) with hierarchy plots
- Gene regulatory networks — Use
grn-pyscenicto find transcription factors driving the communication - Functional enrichment — Run pathway analysis on sender/receiver gene sets
Related Skills
| Skill | Relationship |
|---|---|
scrnaseq-seurat-core-analysis |
Upstream — produces the annotated Seurat object input |
scrnaseq-scanpy-core-analysis |
Alternative upstream (Python-based, convert to Seurat for CellChat) |
grn-pyscenic |
Complementary — gene regulatory networks from same scRNA-seq data |
References
- Jin S, et al. Inference and analysis of cell-cell communication using CellChat. Nature Communications. 2021;12:1088.
- Jin S, et al. CellChat for systematic analysis of cell-cell communication from single-cell and spatially resolved transcriptomics. Nature Protocols. 2024.
- CellChat v2 GitHub (active)
- CellChat tutorials
- Detailed patterns: references/cellchat-guide.md
- Visualization options: references/visualization-guide.md
Code preview
scripts/cellchat_plots.R
# =============================================================================
# Cell-Cell Communication Analysis — Visualization
# =============================================================================
# Generates 6 publication-quality plots from CellChat analysis results.
# Saves PNG + SVG with graceful SVG fallback.
# =============================================================================
suppressPackageStartupMessages({
library(CellChat)
library(ggplot2)
library(ggprism)
})
# Try to load svglite for high-quality SVG (optional)
.has_svglite <- requireNamespace("svglite", quietly = TRUE)
if (.has_svglite) {
suppressPackageStartupMessages(library(svglite))
}
# --- Save helpers -----------------------------------------------------------
#' Save a ggplot object to PNG + SVG
.save_ggplot <- function(plot, base_path, width = 8, height = 6, dpi = 300) {
# Always save PNG
png_path <- sub("\\.(svg|png)$", ".png", base_path)
ggsave(png_path, plot = plot, width = width, height = height, dpi = dpi,
device = "png")
cat(" Saved:", png_path, "\n")
# Always try SVG
svg_path <- sub("\\.(svg|png)$", ".svg", base_path)
tryCatch({
ggsave(svg_path, plot = plot, width = width, height = height,
device = "svg")
cat(" Saved:", svg_path, "\n")
}, error = function(e) {
tryCatch({
svg(svg_path, width = width, height = height)
print(plot)
dev.off()
cat(" Saved:", svg_path, "\n")
}, error = function(e2) {
cat(" (SVG export failed for ggplot)\n")
})
})
}
#' Save a base R plot to PNG + SVG using a plot function (closure)
#' @param plot_fn A zero-argument function that draws the plot
.save_base_plot <- function(plot_fn, base_path, width = 8, height = 6,
dpi = 300) {
# Always save PNG
png_path <- sub("\\.(svg|png)$", ".png", base_path)
png(png_path, width = width, height = height, units = "in", res = dpi)
tryCatch(plot_fn(), error = function(e) {
cat(" ⚠ PNG plot error:", conditionMessage(e), "\n")
})
dev.off()
cat(" Saved:", png_path, "\n")
# Always try SVG
svg_path <- sub("\\.(svg|png)$", ".svg", base_path)
tryCatch({
svg(svg_path, width = width, height = height)
plot_fn()
dev.off()
cat(" Saved:", svg_path, "\n")
}, error = function(e) {
tryCatch(dev.off(), error = function(e2) NULL)
cat(" (SVG export failed for base plot)\n")
})
}
# --- Individual Plot Functions -----------------------------------------------
#' Plot 1: Interaction count network (circle plot)
plot_interaction_count_network <- function(cellchat, output_dir) {
cat("\n [1/6] Interaction count network...\n")
scripts/export_results.R
# =============================================================================
# Cell-Cell Communication Analysis — Export Results
# =============================================================================
# Exports all CellChat analysis results: CSV tables, CellChat RDS object,
# markdown report, and PDF report (via generate_report.R).
# =============================================================================
suppressPackageStartupMessages({
library(CellChat)
})
#' Export all CellChat analysis results
#'
#' @param cellchat CellChat object (from run_cellchat_analysis)
#' @param seurat_obj Original Seurat object (optional, for metadata export)
#' @param output_dir Output directory
export_all <- function(cellchat, seurat_obj = NULL, output_dir = "results") {
cat("\n=== Exporting Results ===\n\n")
dir.create(output_dir, showWarnings = FALSE, recursive = TRUE)
# -------------------------------------------------------------------------
# 1. Significant interactions (all L-R pairs with statistics)
# -------------------------------------------------------------------------
cat("1. Significant interactions...\n")
df_net <- subsetCommunication(cellchat)
write.csv(df_net, file.path(output_dir, "significant_interactions.csv"),
row.names = FALSE)
cat(" Saved:", file.path(output_dir, "significant_interactions.csv"),
"(", nrow(df_net), "interactions )\n")
# -------------------------------------------------------------------------
# 2. Pathway-level summary
# -------------------------------------------------------------------------
cat("2. Pathway-level summary...\n")
df_pathway <- subsetCommunication(cellchat, slot.name = "netP")
write.csv(df_pathway, file.path(output_dir, "pathway_summary.csv"),
row.names = FALSE)
cat(" Saved:", file.path(output_dir, "pathway_summary.csv"),
"(", nrow(df_pathway), "pathway-level interactions )\n")
# -------------------------------------------------------------------------
# 3. Interaction count matrix (cell type × cell type)
# -------------------------------------------------------------------------
cat("3. Interaction count matrix...\n")
count_mat <- cellchat@net$count
write.csv(count_mat, file.path(output_dir, "interaction_count_matrix.csv"))
cat(" Saved:", file.path(output_dir, "interaction_count_matrix.csv"), "\n")
# -------------------------------------------------------------------------
# 4. Interaction strength matrix (cell type × cell type)
# -------------------------------------------------------------------------
cat("4. Interaction strength matrix...\n")
weight_mat <- cellchat@net$weight
write.csv(weight_mat, file.path(output_dir, "interaction_strength_matrix.csv"))
cat(" Saved:", file.path(output_dir, "interaction_strength_matrix.csv"), "\n")
# -------------------------------------------------------------------------
# 5. Signaling roles (centrality scores)
# -------------------------------------------------------------------------
cat("5. Signaling role scores...\n")
tryCatch({
# Extract centrality measures for each pathway
centr_list <- list()
pathways <- cellchat@netP$pathways
for (pw in pathways) {
pw_centr <- tryCatch(
cellchat@netP$centr[[pw]],
error = function(e) NULL
)
if (!is.null(pw_centr)) {
df_pw <- data.frame(
pathway = pw,
cell_type = names(pw_centr$outdeg),
outdeg_sender = pw_centr$outdeg,
indeg_receiver = pw_centr$indeg,
flowbet_mediator = if (!is.null(pw_centr$flowbet)) pw_centr$flowbet else NA,
info_influencer = if (!is.null(pw_centr$info)) pw_centr$info else NA,
stringsAsFactors = FALSEscripts/generate_report.R
# =============================================================================
# Cell-Cell Communication Analysis — PDF Report Generation
# =============================================================================
# Generates a publication-quality PDF report using rmarkdown.
# Falls back gracefully if rmarkdown or tinytex is unavailable.
# =============================================================================
#' Generate PDF report from CellChat analysis
#'
#' @param cellchat CellChat object
#' @param df_net Data frame of significant interactions
#' @param df_pathway Data frame of pathway-level interactions
#' @param output_dir Directory containing plots and for output
generate_report <- function(cellchat, df_net, df_pathway,
output_dir = "results") {
# Check rmarkdown availability
if (!requireNamespace("rmarkdown", quietly = TRUE)) {
cat(" rmarkdown not installed — skipping PDF report\n")
cat(" Install with: install.packages('rmarkdown')\n")
return(invisible(NULL))
}
# Check for PDF rendering capability
has_tinytex <- requireNamespace("tinytex", quietly = TRUE) &&
tinytex::is_tinytex()
has_xelatex <- nchar(Sys.which("xelatex")) > 0
has_pdflatex <- nchar(Sys.which("pdflatex")) > 0
if (!has_tinytex && !has_xelatex && !has_pdflatex) {
cat(" No LaTeX installation found — skipping PDF report\n")
cat(" Install with: tinytex::install_tinytex()\n")
return(invisible(NULL))
}
# Create temporary Rmd file
rmd_path <- file.path(output_dir, "_report.Rmd")
pdf_path <- file.path(output_dir, "analysis_report.pdf")
pathways <- cellchat@netP$pathways
cell_types <- levels(cellchat@idents)
n_interactions <- nrow(df_net)
n_pathways <- length(pathways)
n_celltypes <- length(cell_types)
# Top pathways
top_pathways_text <- ""
if (n_pathways > 0) {
pathway_prob <- cellchat@netP$prob
if (!is.null(pathway_prob) && length(dim(pathway_prob)) == 3) {
pathway_strength <- apply(pathway_prob, 3, sum)
pathway_strength <- sort(pathway_strength, decreasing = TRUE)
top_n <- min(10, length(pathway_strength))
top_pw <- data.frame(
Pathway = names(pathway_strength)[1:top_n],
Strength = round(pathway_strength[1:top_n], 4)
)
top_pathways_text <- knitr::kable(top_pw, format = "pipe")
top_pathways_text <- paste(top_pathways_text, collapse = "\n")
}
}
# Top interactions
top_interactions_text <- ""
if (n_interactions > 0) {
df_top <- df_net[order(df_net$prob, decreasing = TRUE), ]
df_top <- head(df_top, 10)
ti <- data.frame(
Source = df_top$source,
Target = df_top$target,
Interaction = df_top$interaction_name_2,
Pathway = df_top$pathway_name,
Prob = round(df_top$prob, 4)
)
top_interactions_text <- knitr::kable(ti, format = "pipe")
top_interactions_text <- paste(top_interactions_text, collapse = "\n")
}
# Find available plot PNGs
plot_files <- list.files(output_dir, pattern = "\\.png$", full.names = TRUE)Companion files
| Type | Path | Bytes |
|---|---|---|
| Markdown | references/cellchat-guide.md | 5,911 |
| Markdown | references/visualization-guide.md | 5,776 |
| R | scripts/cellchat_plots.R | 11,118 |
| R | scripts/export_results.R | 11,626 |
| R | scripts/generate_report.R | 6,266 |
| R | scripts/load_data.R | 6,734 |
| R | scripts/run_cellchat.R | 6,725 |
| Markdown | SKILL.md | 10,222 |
| JSON | skill.meta.json | 1,891 |