Meta: Create a Skill
How to build, test, package and present a reusable skill.
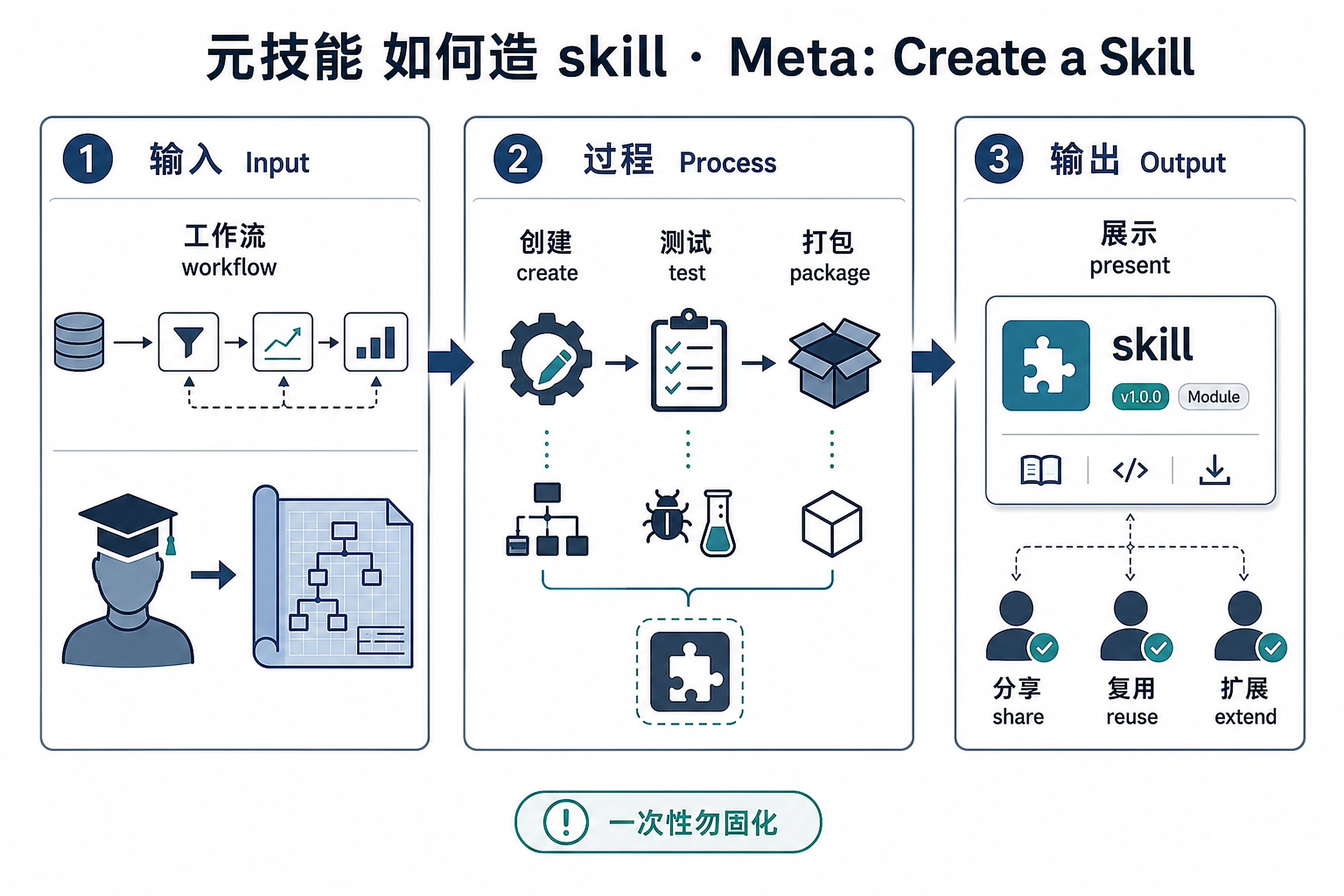
Overview
Problem. After learning all these, how do you author your own?
Learning goals
- Graduate from using skills to authoring them
- Knowing the structure makes you a better reader too
Figures
Tutorial
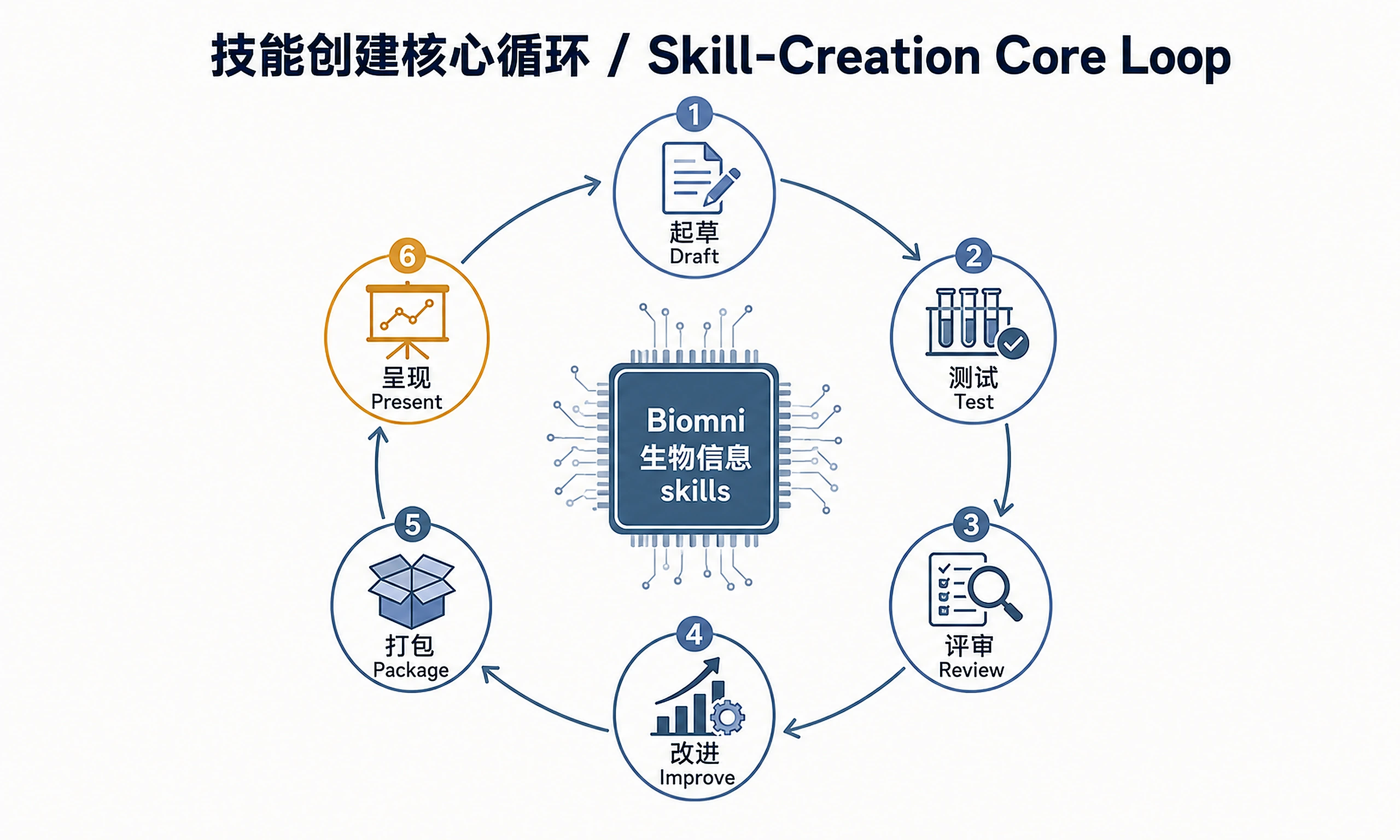
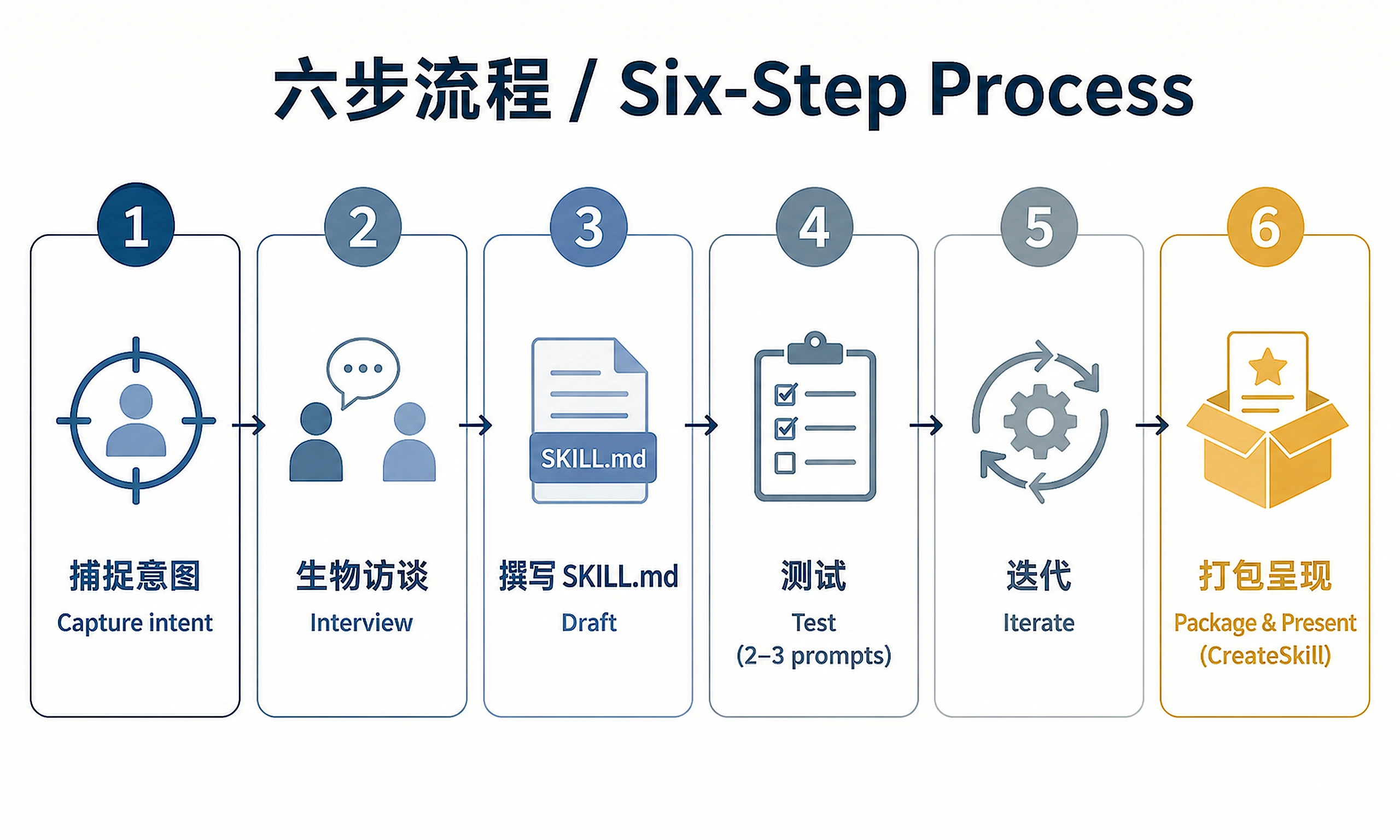
A skill-creator tailored for Phylo's Biomni platform and the bioinformatics domain, with biology-specific interviewing, domain patterns, and eval scaffolding baked in. The core loop is: draft → test → review → improve → package → present.
Process Overview
- Capture intent — understand what the skill does and when it should trigger
- Interview — ask bio-specific clarifying questions (databases, file formats, agent vs. standalone)
- Draft SKILL.md — write the skill instructions and frontmatter
- Test — run 2–3 representative prompts and review output quality
- Iterate — revise based on gaps or mismatches
- Package & present — create the skill folder under
/mnt/results/skills/<slug>/and callCreateSkill
You don't need to go in strict order. If the user already has a draft, jump to testing. If they just want to vibe without evals, that's fine too.
If the user did not explicitly ask you to create a skill, ask first before creating one. Offer skill creation when the workflow is genuinely reusable:
- repeated or likely to recur
- procedural, with stable steps
- worth saving as knowledge for future tasks
Step 1: Capture Intent
Extract from the conversation first — tools used, corrections made, input/output formats observed. Then confirm:
- What should this skill enable Biomni to do?
- What kind of user prompt triggers it? (bioinformatics jargon, file uploads, database names, etc.)
- Does it run inside a Biomni agent session (E2B sandbox, tool call) or standalone?
- What's the expected output — a file, a report, a protocol, code, a database query result?
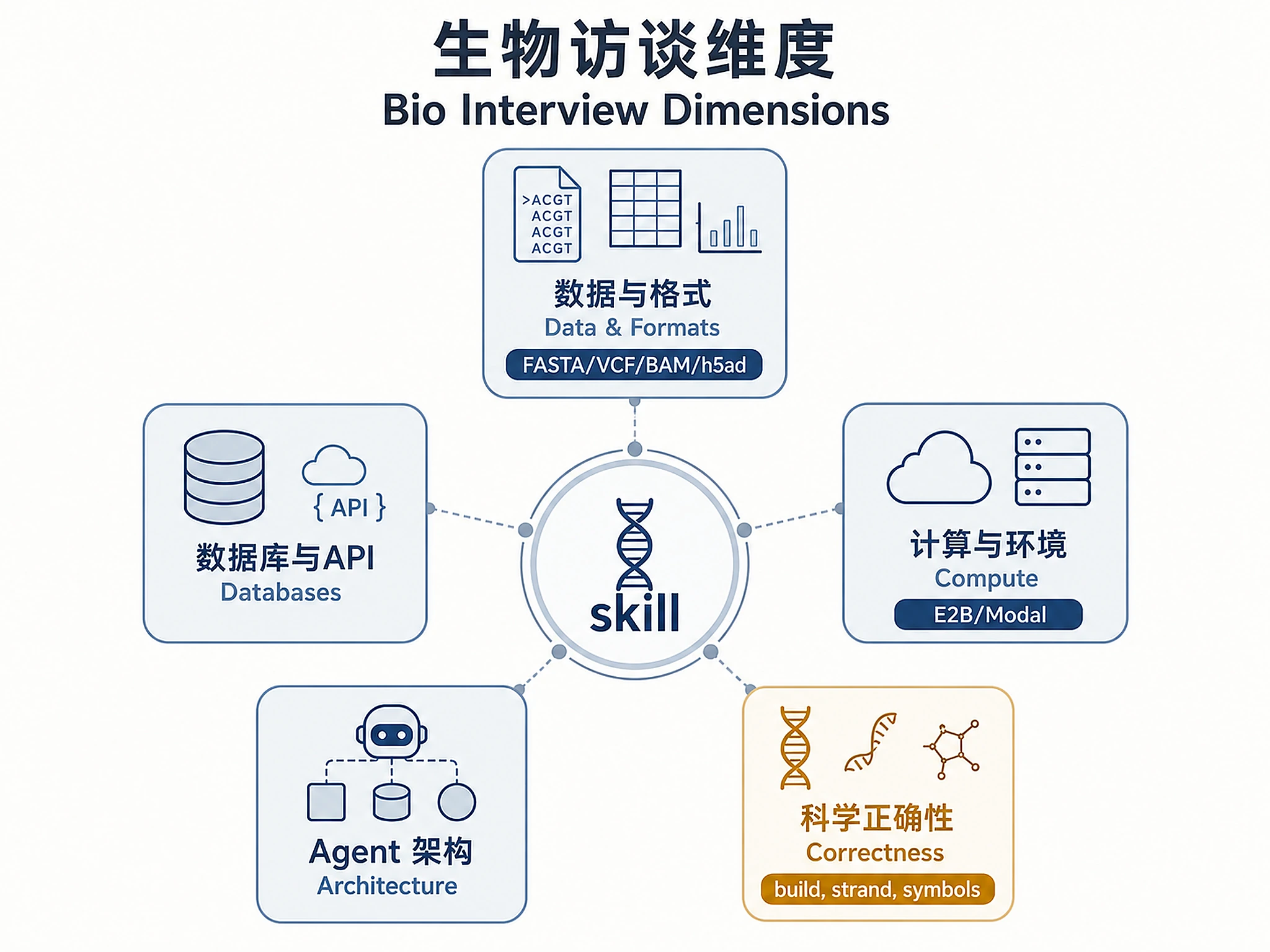
Step 2: Bio-Specific Interview
Ask targeted questions before drafting. Typical gaps in bioinformatics protocols:
Data & Formats
- What file formats are involved? (FASTA/FASTQ, VCF, BAM/SAM, BED, GTF, AnnData
.h5ad, CSV, JSON, PDF protocol) - What's the expected input size? (a handful of gene names vs. a whole-genome VCF)
- Does the skill need to handle multi-sample or batch inputs?
Databases & APIs
- Which biological databases does the skill query?
- Are there licensing or access constraints on any databases (e.g., OMIM, DisGeNET, DepMap require licenses)?
- Does it need to cross-reference multiple databases and reconcile identifiers (gene symbol → Ensembl ID → UniProt accession)?
Compute & Environment
- Does the skill run heavy computation (alignment, docking, ML inference) that needs E2B sandbox resources?
- Does it need Modal or another async runner for long jobs?
- Does it produce files the user downloads (BAM, ZIP of results, PDF report)?
Agent Architecture (if Biomni-facing)
- Is this a new Biomni tool (defined in the A2 agent's tool registry)?
- Does it need to write back to the user's session storage (S3, Firestore)?
- Should it produce SSE streaming output or a single blocking result?
Scientific Correctness
- Are there common failure modes that would silently produce wrong biology? (e.g., ignoring strand, mixing GRCh37/GRCh38 coordinates, using non-canonical gene symbols)
- Should the skill sanity-check its own outputs before returning them?
Step 3: Write the SKILL.md
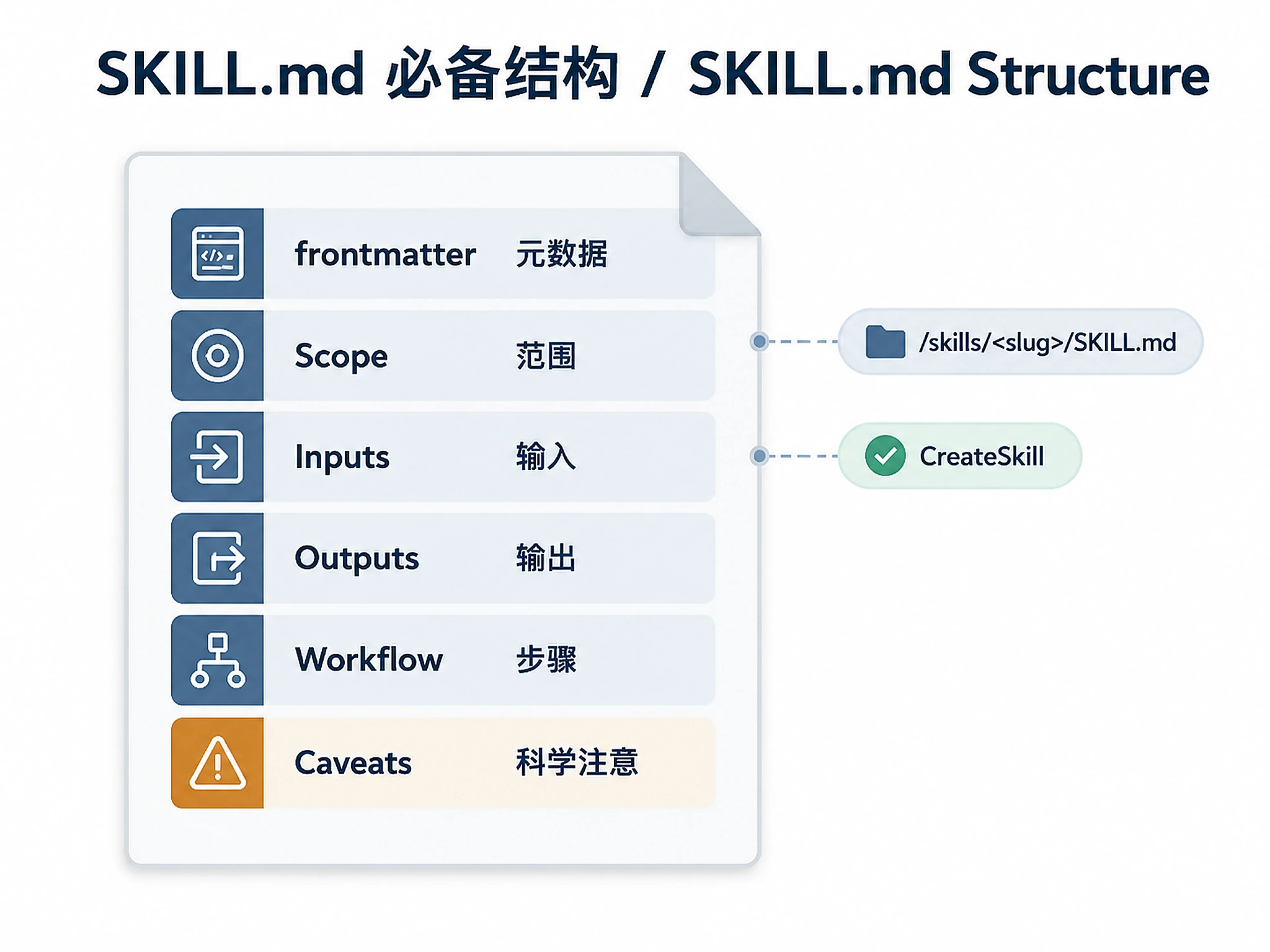
Every bioinformatics protocol SKILL.md should include:
Required sections
- YAML frontmatter —
name,description(include biology keywords that would trigger it) - Scope — one sentence on what it does and what it explicitly does NOT do
- Inputs — file formats, identifier types, expected ranges
- Outputs — format, where it's saved (S3 path / local / chat)
- Workflow steps — numbered, imperative, with biological context for why each step matters
- Scientific caveats — genome build assumptions, known edge cases, licensing flags
When generating a reusable skill package, the top-level file must live at:
/mnt/results/skills/<slug>/SKILL.md
Put any supporting files in the same folder tree. Do not write generated skill packages at the root of /mnt/results.
Optional sections (add when relevant)
- Database reference table — which DBs are queried, what identifiers they accept, rate limits
- File format handling — how to parse/validate the format before processing
- Error handling — what to do when a gene isn't found, a database is down, coordinates are out of range
Description writing tips for biology skills
The description must contain enough domain vocabulary to trigger reliably. Include:
- The names of specific databases, tools, or file formats involved
- The scientific task type (variant annotation, pathway analysis, docking, protocol generation, etc.)
- The Biomni context if applicable ("Biomni tool", "agent session", "lab automation")
Make descriptions slightly pushy: instead of "Annotates VCF files", write "Annotates VCF files with ClinVar, gnomAD, and COSMIC data. Use this skill whenever someone uploads a VCF, asks about variant pathogenicity, or wants to know if a mutation is in a cancer database — even if they don't say 'annotate'."
Step 4: Test Cases
After drafting, propose 2–3 test prompts that mirror real user requests. Good bioinformatics protocol test prompts are:
- Specific enough to trigger the skill ("Annotate this VCF with ClinVar data" not "help me with variants")
- Biologically realistic — use real gene names, real database names, plausible file names
- Representative of the range — one simple case, one with a tricky edge (multi-sample, missing ID, big file), one that could go wrong scientifically
Share test prompts with the user before running: "Here are three prompts I'd like to test. Do they match what real Biomni users would say?"
Run each one manually (since there are no subagents — do them sequentially). For each run:
- Follow the skill's instructions as if encountering the task fresh
- Record: did it produce the right output? Did it catch biological errors? Was the output format correct?
Step 5: Evaluate & Iterate
What to look for in bioinformatics protocol outputs
Scientific correctness
- Are gene/protein identifiers resolved consistently?
- Are genome coordinates in the right build?
- Are database citations accurate (no hallucinated PMIDs, no made-up ClinVar accessions)?
Completeness
- Does the output include all the fields the user needs?
- Are edge cases (no results found, ambiguous gene name, deprecated ID) handled gracefully?
Format fidelity
- If the skill outputs a file (FASTA, VCF, report PDF), is it properly formatted?
- If it outputs a protocol, does it follow a standard lab format?
Biomni integration (if applicable)
- Does the skill correctly reference Biomni tool schemas?
- Does it produce the right SSE event structure?
- Are S3 paths and session IDs handled correctly?
Ask the user to review outputs and flag:
- Any scientifically wrong result (even subtle errors matter in biology)
- Missing context a researcher would expect
- Formatting that doesn't match what Biomni renders
Revise the skill and re-run. Repeat until you're both happy with it.
Step 6: Package & Present
Once satisfied, create the generated skill package under:
/mnt/results/skills/<slug>/
The package must include:
SKILL.mdat the folder root- YAML frontmatter with at least
nameanddescription - concise instructions focused on when to use the skill and how to run the workflow
After the package is written, call the CreateSkill tool with:
{
"folder_paths": ["skills/<slug>"]
}
Call CreateSkill as the final action. The tool validates the generated skill, surfaces the preview UI for SKILL.md, and lets the user add the skill to personal skills.
Code preview
No Python/R preview files were found.
Companion files
| Type | Path | Bytes |
|---|---|---|
| Markdown | SKILL.md | 7,707 |
| JSON | skill.meta.json | 588 |