Phylogenetics Toolkit
End-to-end: align → tree → ancestral reconstruction → clock.
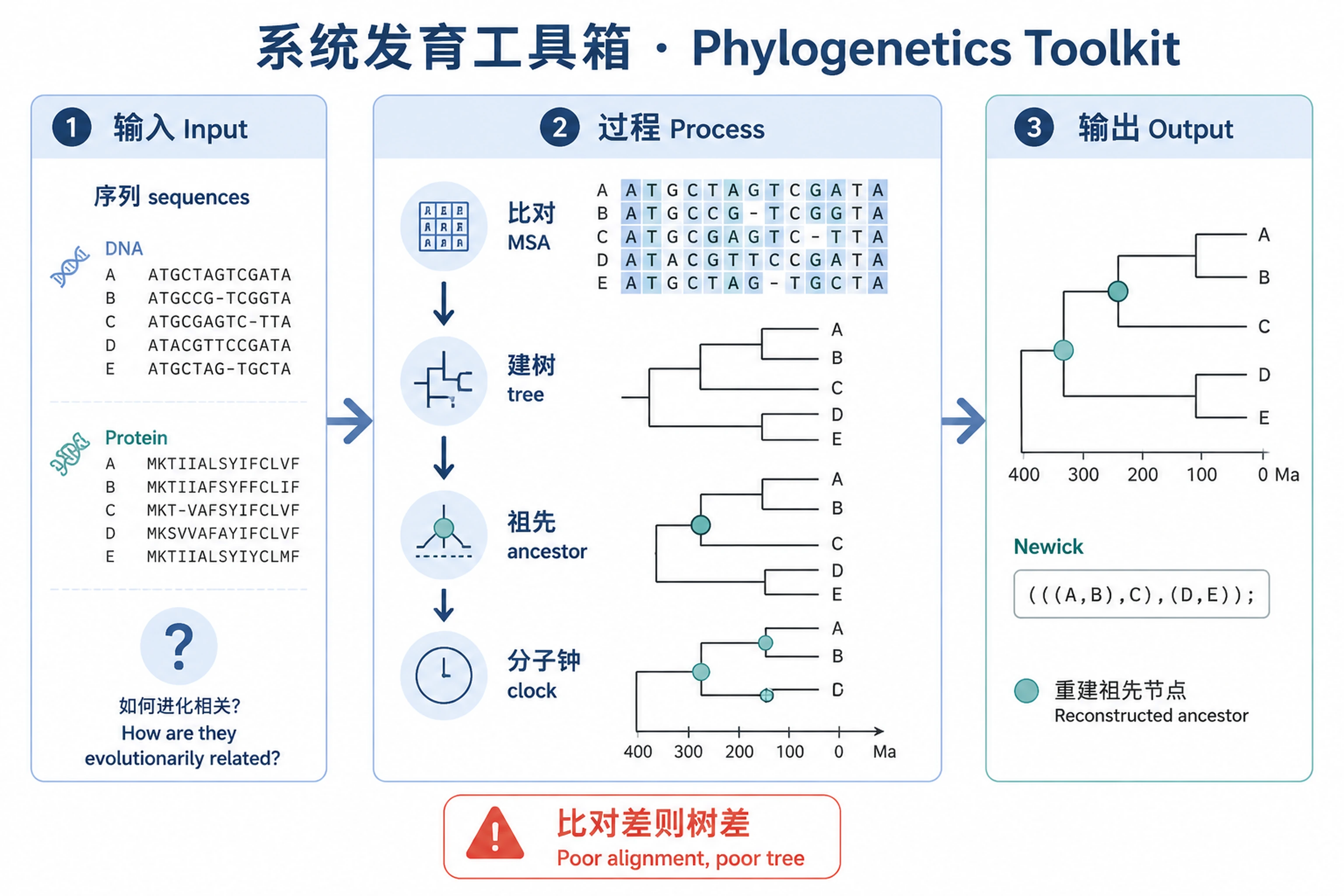
Overview
Problem. How are these sequences related; what was the ancestor?
Learning goals
- Alignment quality determines tree quality
- Newick is the standard tree text format
Figures
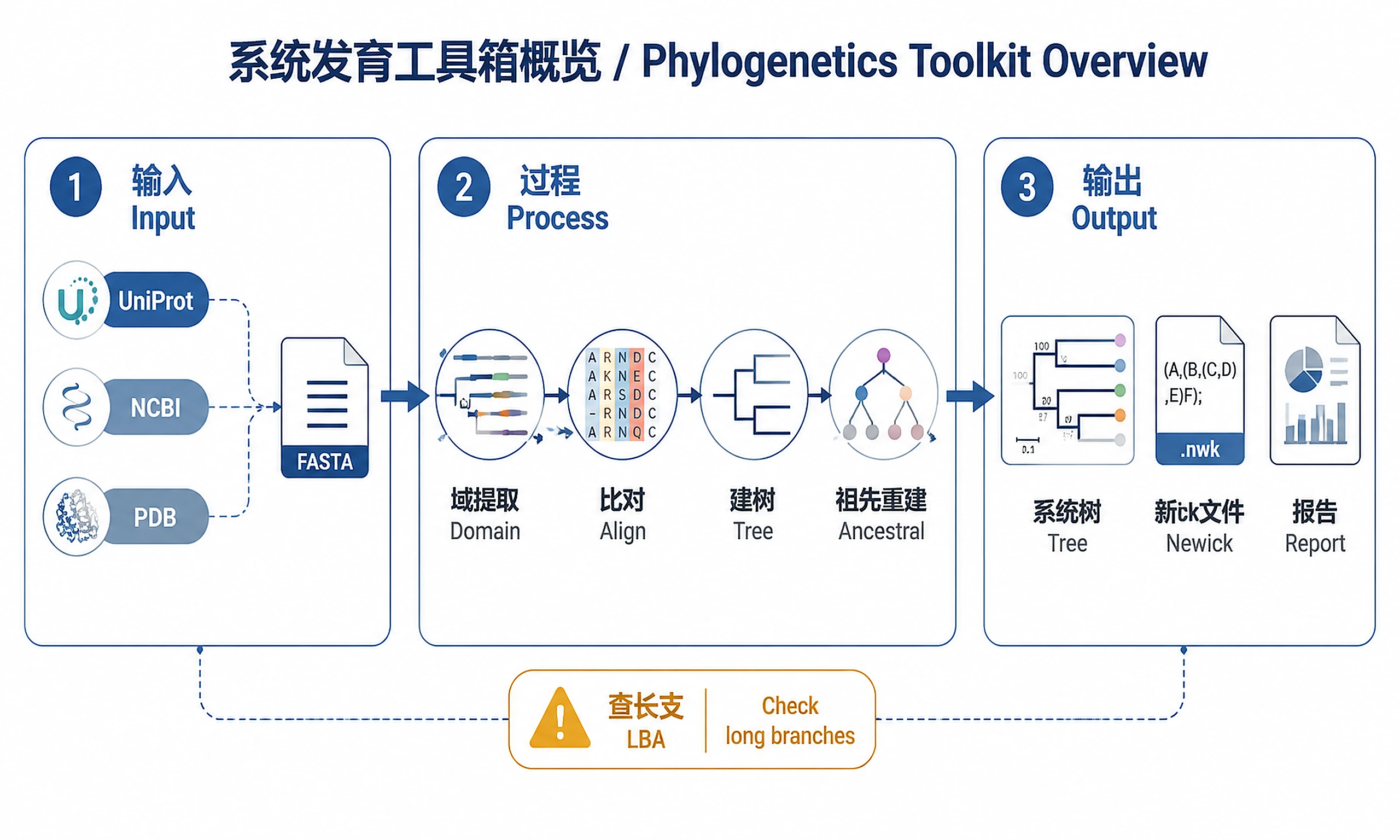
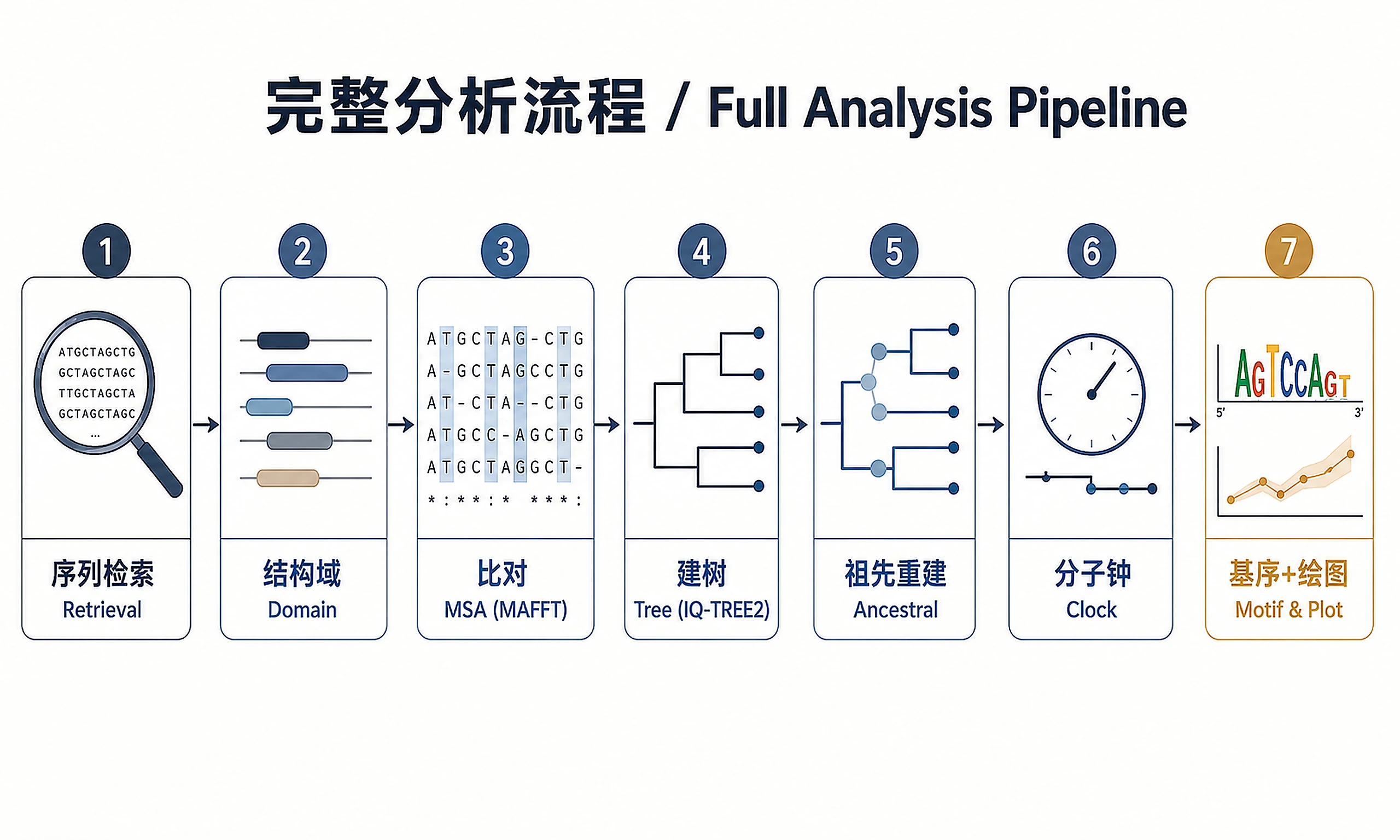
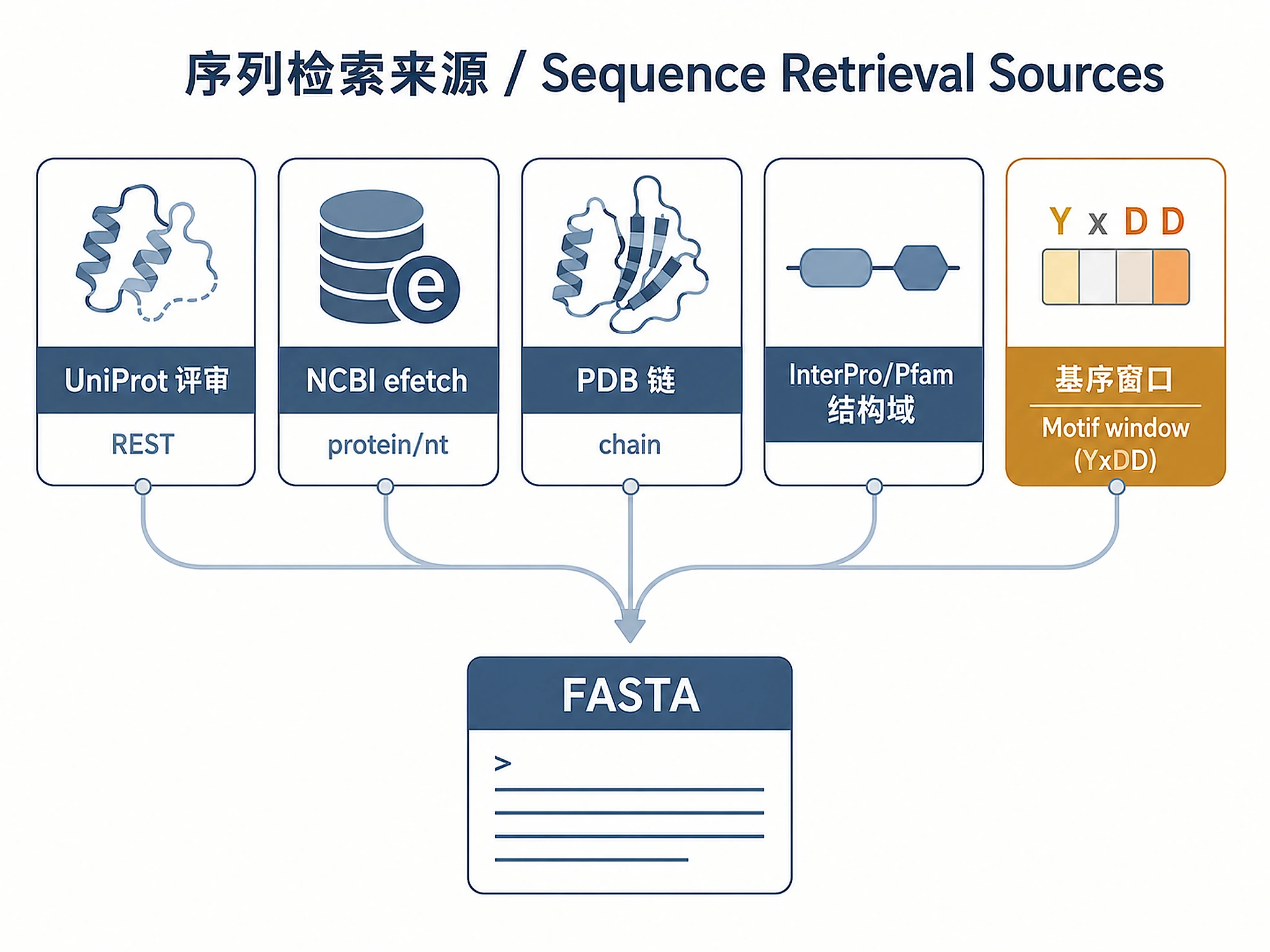
Tutorial
A unified workflow for protein (or nucleotide) phylogenetic analysis, from raw sequence identifiers to publication-ready figures. All steps are implemented in Python unless noted. Validated on divergent protein families (EC 2.7.7.*, RT palm domain, ~20–350 sequences, 10–40% pairwise identity).
0. Sanity-check before starting
Before running any step, confirm:
- Sequence source: UniProt accessions, NCBI accessions, PDB chains, or FASTA file?
- Domain scope: Full-length or specific domain (e.g., RT palm, Pfam domain)?
- Outgroup: Is there a clear outgroup (non-homologous fold, or known basal lineage)?
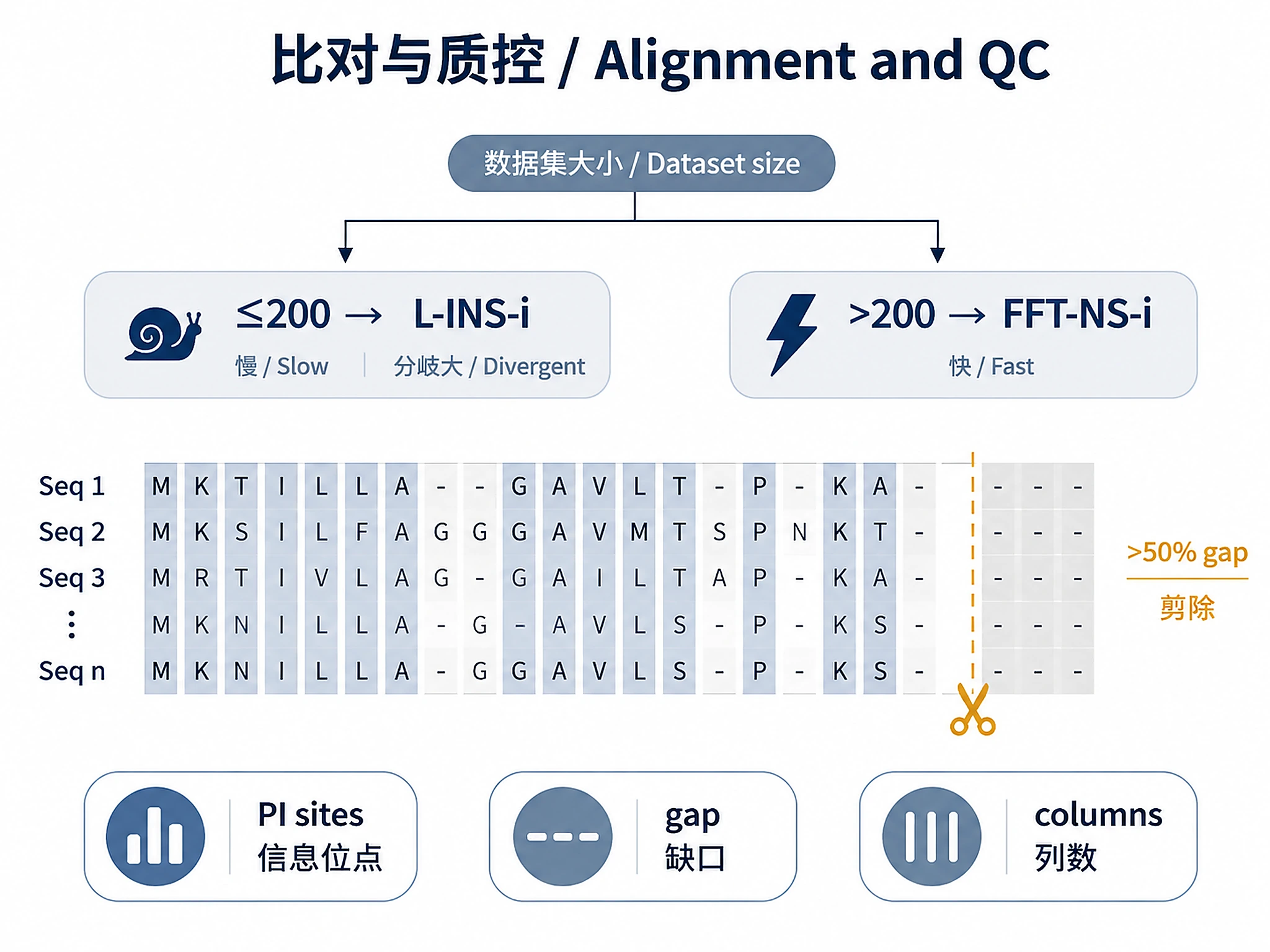
- Dataset size: ≤200 sequences → L-INS-i; >200 → FFT-NS-i.
- Calibration points: Are tip dates available for molecular clock? If not, use relative dating.
1. Sequence Retrieval
1a. UniProt REST API (Swiss-Prot reviewed)
import requests, re
def fetch_uniprot(query: str, format: str = "fasta", reviewed: bool = True) -> str:
"""
query: UniProt query string, e.g. 'ec:2.7.7.* AND reviewed:true'
Returns: FASTA string
"""
base = "https://rest.uniprot.org/uniprotkb/stream"
params = {
"query": query + (" AND reviewed:true" if reviewed else ""),
"format": format,
"compressed": "false"
}
r = requests.get(base, params=params, timeout=120)
r.raise_for_status()
return r.text
# Example: fetch all Swiss-Prot EC 2.7.7.* sequences
fasta = fetch_uniprot("ec:2.7.7.*")
# Example: fetch specific accessions
def fetch_uniprot_accessions(accessions: list[str]) -> str:
ids = " OR ".join(f"accession:{a}" for a in accessions)
return fetch_uniprot(ids, reviewed=False)
1b. NCBI efetch (protein or nucleotide)
from Bio import Entrez, SeqIO
import io
Entrez.email = "your@email.com"
def fetch_ncbi_protein(accessions: list[str]) -> list:
"""Fetch protein sequences from NCBI by accession list."""
handle = Entrez.efetch(
db="protein",
id=",".join(accessions),
rettype="fasta",
retmode="text"
)
records = list(SeqIO.parse(handle, "fasta"))
handle.close()
return records
# Example
records = fetch_ncbi_protein(["WP_211092143.1", "NP_057856.1"])
1c. PDB chain extraction
import requests
def fetch_pdb_fasta(pdb_id: str, chain: str = None) -> str:
"""Fetch FASTA for a PDB entry (all chains or specific chain)."""
url = f"https://www.rcsb.org/fasta/entry/{pdb_id.upper()}"
r = requests.get(url, timeout=30)
r.raise_for_status()
if chain is None:
return r.text
# Filter to specific chain
lines = r.text.split("\n")
result, keep = [], False
for line in lines:
if line.startswith(">"):
keep = f"|Chain{chain}|" in line or f"Chain {chain}" in line
if keep:
result.append(line)
return "\n".join(result)
# Example: Drt3a (chain A) and Drt3b (chain G) from PDB 9Z6Z
fasta_drt3a = fetch_pdb_fasta("9Z6Z", "A")
fasta_drt3b = fetch_pdb_fasta("9Z6Z", "G")
1d. Domain extraction via InterPro/Pfam
import requests, time
def get_pfam_coordinates(uniprot_acc: str, pfam_id: str = None) -> list[dict]:
"""
Returns list of {pfam_id, start, end, name} for a UniProt accession.
If pfam_id given, filters to that domain only.
"""
url = f"https://www.ebi.ac.uk/interpro/api/entry/pfam/protein/UniProt/{uniprot_acc}/?format=json"
r = requests.get(url, timeout=30)
if r.status_code != 200:
return []
data = r.json()
results = []
for entry in data.get("results", []):
acc = entry["metadata"]["accession"]
name = entry["metadata"]["name"]
for loc in entry.get("proteins", [{}])[0].get("entry_protein_locations", []):
for frag in loc.get("fragments", []):
if pfam_id is None or acc == pfam_id:
results.append({"pfam_id": acc, "name": name,
"start": frag["start"], "end": frag["end"]})
time.sleep(0.2) # rate limit
return results
def trim_sequence_to_domain(seq: str, start: int, end: int, padding: int = 0) -> str:
"""Extract domain region (1-indexed, inclusive). Optional padding."""
s = max(0, start - 1 - padding)
e = min(len(seq), end + padding)
return seq[s:e]
1e. Motif-based domain extraction (fallback for unannotated sequences)
import re
def find_motif_window(seq: str, motif_regex: str = r"Y[A-Z]DD",
upstream: int = 100, downstream: int = 100) -> tuple[str, int]:
"""
Extract a window around a conserved motif (e.g., YXDD for RT palm).
Returns (subsequence, motif_start_position).
"""
m = re.search(motif_regex, seq)
if m is None:
return None, -1
pos = m.start()
start = max(0, pos - upstream)
end = min(len(seq), pos + downstream)
return seq[start:end], pos
# Example: extract RT palm domain around YXDD
palm_seq, yxdd_pos = find_motif_window(full_seq, r"Y[A-Z]DD", upstream=100, downstream=100)
2. Multiple Sequence Alignment
2a. MAFFT (recommended)
import subprocess, tempfile, os
from Bio import SeqIO
import io
def run_mafft(sequences: dict[str, str], algorithm: str = "linsi",
n_threads: int = 4) -> dict[str, str]:
"""
sequences: {name: sequence} dict
algorithm: 'linsi' (≤200 seqs, divergent) or 'fftns' (>200 seqs)
Returns: aligned {name: aligned_sequence} dict
"""
# Write input FASTA
with tempfile.NamedTemporaryFile(mode="w", suffix=".fasta", delete=False) as f:
for name, seq in sequences.items():
f.write(f">{name}\n{seq}\n")
infile = f.name
outfile = infile.replace(".fasta", "_aligned.fasta")
algo_flag = "--localpair --maxiterate 1000" if algorithm == "linsi" else "--retree 2 --reorder"
cmd = f"mafft {algo_flag} --thread {n_threads} --quiet {infile} > {outfile}"
subprocess.run(cmd, shell=True, check=True)
aligned = {}
for rec in SeqIO.parse(outfile, "fasta"):
aligned[rec.id] = str(rec.seq)
os.unlink(infile)
os.unlink(outfile)
return aligned
# Decision rule:
# - ≤200 sequences, divergent (e.g., <40% mean identity): use 'linsi'
# - >200 sequences or fast preview needed: use 'fftns'
2b. Gap trimming
import numpy as np
def trim_alignment_by_gap(aligned: dict[str, str], max_gap_frac: float = 0.5) -> dict[str, str]:
"""
Remove alignment columns where gap fraction > max_gap_frac.
Default: remove columns with >50% gaps.
"""
names = list(aligned.keys())
seqs = [list(aligned[n]) for n in names]
n_seqs = len(seqs)
n_cols = len(seqs[0])
keep_cols = []
for col in range(n_cols):
gap_count = sum(1 for s in seqs if s[col] == "-")
if gap_count / n_seqs <= max_gap_frac:
keep_cols.append(col)
trimmed = {}
for i, name in enumerate(names):
trimmed[name] = "".join(seqs[i][col] for col in keep_cols)
return trimmed
# Alternative: trimAl (if installed)
# subprocess.run(f"trimal -in aligned.fasta -out trimmed.fasta -automated1", shell=True)
2c. Alignment quality check
def alignment_quality(aligned: dict[str, str]) -> dict:
"""Report alignment statistics."""
seqs = list(aligned.values())
n_seqs = len(seqs)
n_cols = len(seqs[0])
# Parsimony-informative sites
pi_sites = 0
for col in range(n_cols):
chars = [s[col] for s in seqs if s[col] != "-"]
counts = {}
for c in chars:
counts[c] = counts.get(c, 0) + 1
# PI site: ≥2 different characters, each present in ≥2 sequences
variable = {c: v for c, v in counts.items() if v >= 2}
if len(variable) >= 2:
pi_sites += 1
gap_content = sum(s.count("-") for s in seqs) / (n_seqs * n_cols)
return {
"n_sequences": n_seqs,
"n_columns": n_cols,
"parsimony_informative_sites": pi_sites,
"pi_fraction": pi_sites / n_cols,
"gap_content": gap_content
}
# Thresholds:
# pi_fraction > 0.5 → good signal
# gap_content > 0.4 → consider stricter trimming
# n_columns < 50 → alignment too short; expand domain window
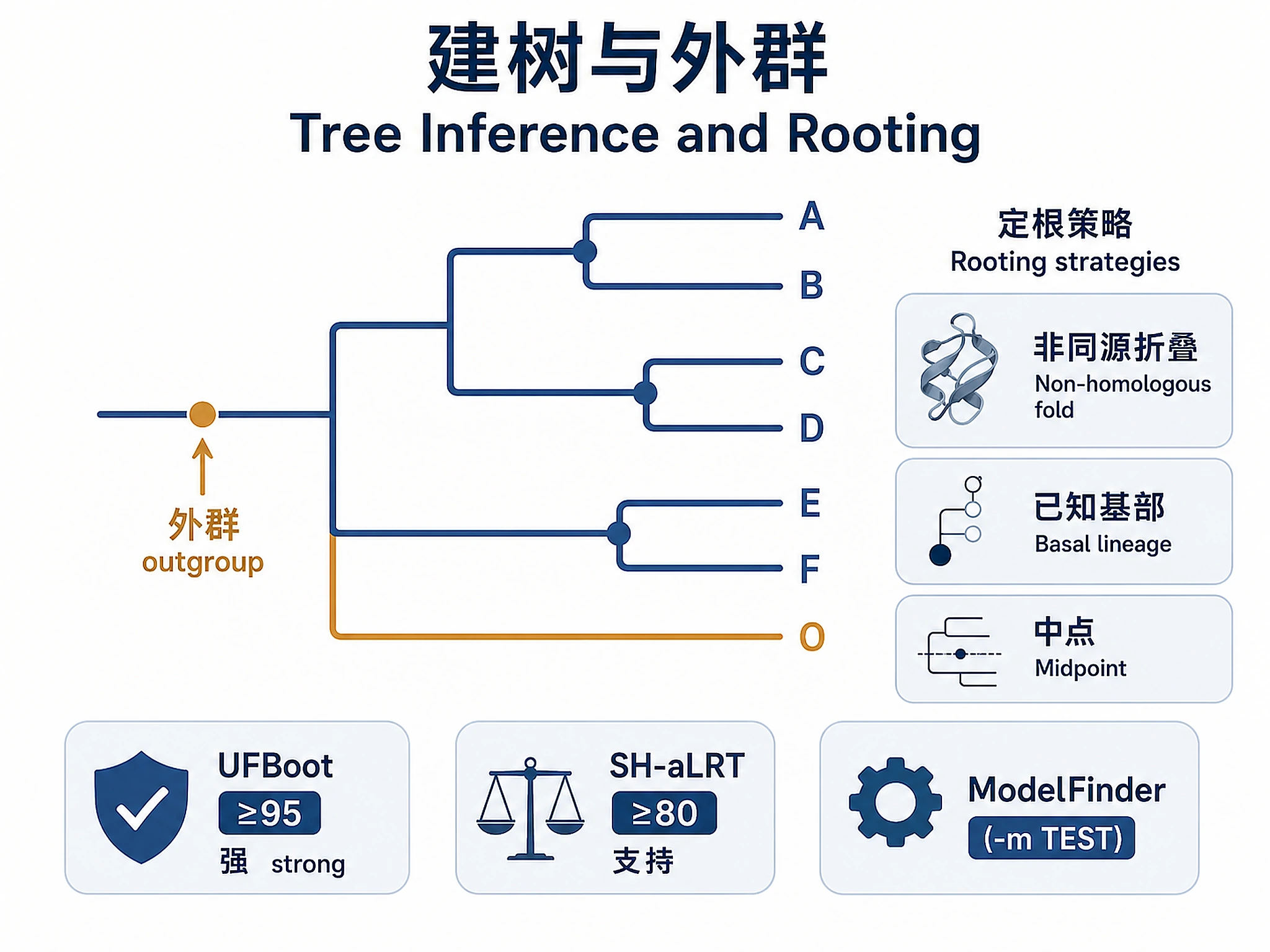
3. Phylogenetic Tree Inference (IQ-TREE2)
3a. Standard run
import subprocess, os
def run_iqtree2(alignment_fasta: str, prefix: str, outgroup: str = None,
n_threads: int = 4, bootstrap: int = 1000) -> str:
"""
alignment_fasta: path to trimmed FASTA alignment
prefix: output file prefix
outgroup: taxon name for rooting (must match FASTA header exactly)
Returns: path to .treefile
"""
cmd = [
"iqtree2", "-s", alignment_fasta,
"--prefix", prefix,
"-m", "TEST", # ModelFinder
"-bb", str(bootstrap), # ultrafast bootstrap
"-alrt", str(bootstrap),# SH-aLRT
"-T", str(n_threads),
"--quiet"
]
if outgroup:
cmd += ["-o", outgroup]
# For small datasets (<50 taxa), use standard bootstrap instead:
# cmd = [c for c in cmd if c not in ["-bb", str(bootstrap)]]
# cmd += ["-b", "100"]
subprocess.run(cmd, check=True)
return f"{prefix}.treefile"
# IMPORTANT: For datasets <50 taxa, UFBoot values are inflated.
# Use -b 100 (standard bootstrap) instead of -bb 1000.
# UFBoot ≥95 = strong support; ≥70 = moderate; SH-aLRT ≥80 = supported.
3b. Model selection guidance
| Dataset type | Expected best model |
|---|---|
| Divergent proteins (e.g., polymerases) | LG+I+G4 or LG+G4 |
| NTase/RNAP families | Q.pfam+I+G4 or Q.yeast+G4 |
| Viral proteins | LG+F+I+G4 |
| Nucleotide sequences | GTR+I+G4 |
Always use -m TEST (ModelFinder) and let IQ-TREE2 select. Do not hard-code the model.
3c. Outgroup selection strategy
- Non-homologous fold: Use a protein with the same reaction chemistry but a different fold (e.g., RdRp as outgroup for RT phylogeny — both are right-hand palm polymerases but RdRp is not an RT).
- Known basal lineage: Use the most ancient lineage as outgroup (e.g., Group II intron RT for retroviral RT phylogeny).
- Midpoint rooting: Use when no outgroup is available (
--root-midpointin IQ-TREE2 or ETE3tree.set_outgroup(tree.get_midpoint_outgroup())).
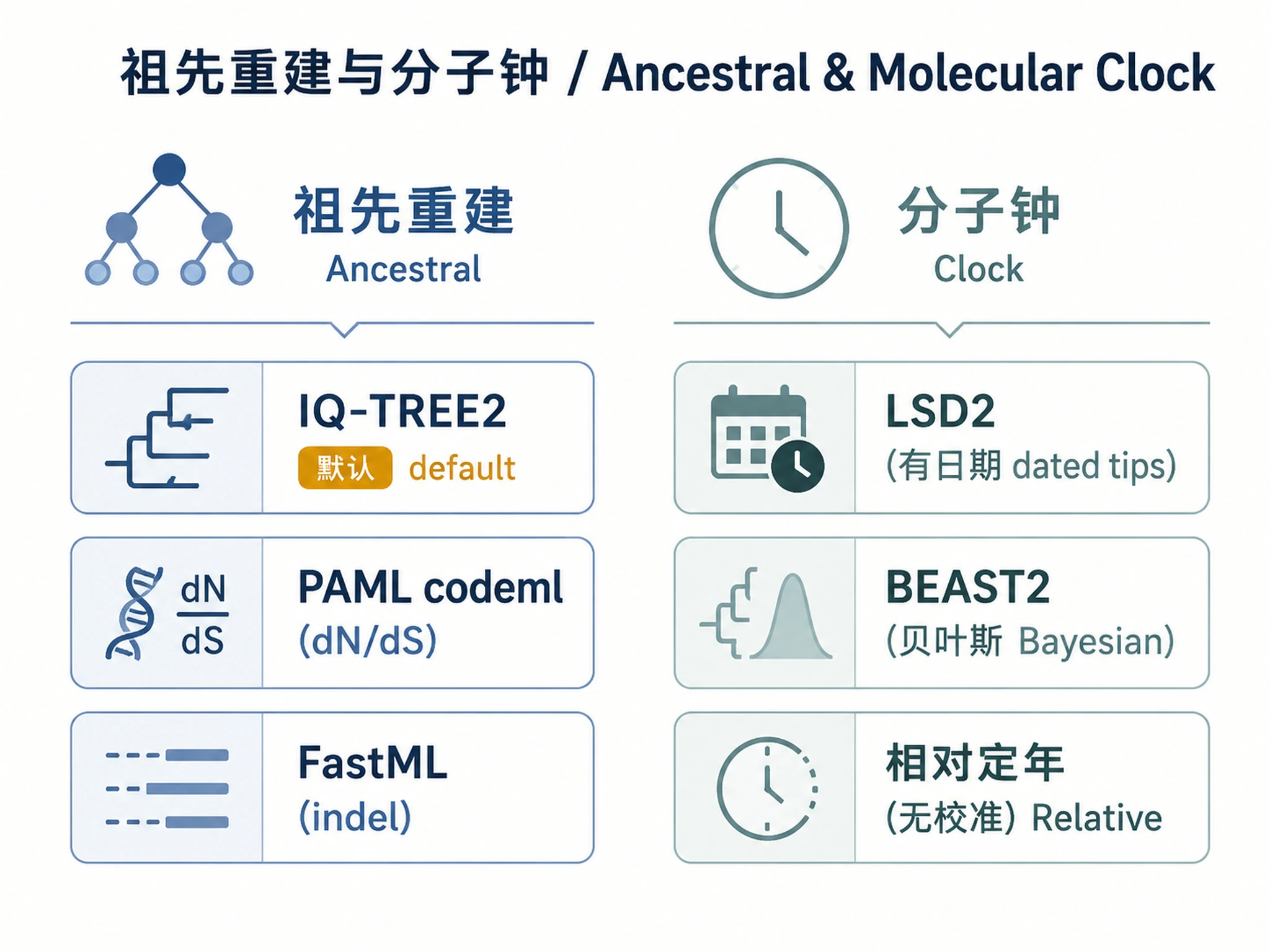
4. Ancestral Sequence Reconstruction
4a. IQ-TREE2 marginal reconstruction (default — fast, integrated)
def run_iqtree2_asr(alignment_fasta: str, treefile: str, prefix: str,
model: str = "LG+I+G4", n_threads: int = 4) -> str:
"""
Marginal ancestral reconstruction using IQ-TREE2.
Returns path to .state file (posterior probabilities per node per site).
"""
cmd = [
"iqtree2", "-s", alignment_fasta,
"-te", treefile, # use fixed tree topology
"-m", model, # use best-fit model from tree inference step
"--ancestral", # marginal reconstruction
"--prefix", prefix + "_asr",
"-T", str(n_threads),
"--quiet"
]
subprocess.run(cmd, check=True)
return f"{prefix}_asr.state"
# Output files:
# .state → posterior probability of each amino acid at each internal node
# .asr.fa → most likely ancestral sequences (FASTA)
# Use the .state file for uncertainty quantification
4b. PAML (codeml) — for dN/dS analysis
# Use when: you need dN/dS ratios, branch-specific selection tests, or codon models
# Requires: codon-aligned nucleotide sequences (not protein)
# Install: conda install -c bioconda paml
# Control file template (write to codeml.ctl):
PAML_CTL = """
seqfile = alignment.phy
treefile = tree.nwk
outfile = codeml_output.txt
noisy = 0
verbose = 0
runmode = 0 * 0: user tree
seqtype = 1 * 1: codons
CodonFreq = 2 * F3x4
model = 0 * 0: one omega for all branches
NSsites = 0 * 0: M0 (one ratio)
icode = 0 * universal genetic code
fix_kappa = 0
kappa = 2
fix_omega = 0
omega = 0.5
"""
4c. FastML — protein ancestral reconstruction
# Use when: protein sequences, need fast marginal reconstruction with indel handling
# Web server: http://fastml.tau.ac.il/
# CLI: fastml -s alignment.fasta -t tree.nwk -m LG -x -y
# Python wrapper:
def run_fastml(alignment_fasta: str, treefile: str, prefix: str,
model: str = "LG") -> None:
cmd = f"fastml -s {alignment_fasta} -t {treefile} -m {model} -x -y -d {prefix}_fastml/"
subprocess.run(cmd, shell=True, check=True)
Decision rule for ancestral reconstruction:
- Default: IQ-TREE2
--ancestral(fast, no extra install, integrated with tree inference) - Need dN/dS or codon-level analysis: PAML codeml
- Need indel reconstruction or web-based: FastML
- Need full Bayesian posterior: BEAST2 (see Section 5b)
5. Molecular Clock Analysis
5a. LSD2 (integrated in IQ-TREE2) — for dated tips
def run_lsd2(alignment_fasta: str, treefile: str, dates_file: str,
prefix: str, n_threads: int = 4) -> str:
"""
dates_file: tab-separated file with taxon_name<TAB>date (e.g., 2020.5)
Returns: path to dated tree
"""
cmd = [
"iqtree2", "-s", alignment_fasta,
"-te", treefile,
"--date", dates_file,
"--date-ci", "100", # 100 bootstrap replicates for CI
"--prefix", prefix + "_dated",
"-T", str(n_threads),
"--quiet"
]
subprocess.run(cmd, check=True)
return f"{prefix}_dated.timetree.nwk"
5b. BEAST2 — full Bayesian relaxed clock
# Use when: tip dates available, need full posterior distribution on divergence times
# Install: conda install -c bioconda beast2
# Requires: BEAUti GUI or XML template for model specification
# Typical run: beast -threads 4 -seed 12345 beast_input.xml
# Post-processing: TreeAnnotator to summarize posterior trees
# Visualization: FigTree or ggtree (R)
5c. Relative dating (no calibration points)
# When no tip dates or fossil calibrations are available:
# Use branch lengths (substitutions/site) as a proxy for relative age.
# Longer branches = more substitutions = either older divergence OR faster evolution.
# ALWAYS check for long-branch attraction (LBA) before interpreting branch lengths.
def flag_long_branches(treefile: str, threshold_multiplier: float = 3.0) -> list[str]:
"""Flag taxa with branch length > threshold_multiplier × mean branch length."""
from Bio import Phylo
import io
tree = Phylo.read(treefile, "newick")
lengths = [c.branch_length for c in tree.find_clades() if c.branch_length]
mean_bl = sum(lengths) / len(lengths)
threshold = threshold_multiplier * mean_bl
flagged = [c.name for c in tree.get_terminals()
if c.branch_length and c.branch_length > threshold]
return flagged, mean_bl, threshold
6. Motif Matching
6a. Standard polymerase palm motifs
import re
PALM_MOTIFS = {
"motif_A": r"[DE]x{1,3}[LIVMF]x{1,3}[DE]", # DxD or ExE variant
"motif_B": r"LP[A-Z]G", # LPXG (RT/RdRp)
"motif_C": r"Y[A-Z]DD", # YXDD (RT palm)
"motif_C_broad": r"[YF][A-Z][DE][DE]", # broader motif C
"GDD": r"GDD", # RdRp motif C
"YGDD": r"YGDD", # RdRp motif C strict
}
def find_all_motifs(seq: str, motifs: dict = PALM_MOTIFS) -> dict[str, list[tuple]]:
"""
Returns {motif_name: [(start, end, matched_string), ...]} for all motifs found.
Positions are 1-indexed.
"""
results = {}
for name, pattern in motifs.items():
matches = [(m.start() + 1, m.end(), m.group())
for m in re.finditer(pattern, seq)]
if matches:
results[name] = matches
return results
# Example
motifs_found = find_all_motifs(drt3a_seq)
# {'motif_B': [(219, 223, 'LPPG')], 'motif_C': [(194, 198, 'YVDD')]}
6b. HMMER domain search
import subprocess, tempfile
def run_hmmsearch(sequences_fasta: str, hmm_db: str = None,
pfam_db: str = "/mnt/shared-workspace/Pfam-A.hmm",
e_threshold: float = 1e-3) -> str:
"""
Run hmmsearch against Pfam or custom HMM database.
Returns path to output table.
"""
outfile = sequences_fasta.replace(".fasta", "_hmmsearch.tbl")
db = hmm_db or pfam_db
cmd = f"hmmsearch --tblout {outfile} -E {e_threshold} --cpu 4 {db} {sequences_fasta}"
subprocess.run(cmd, shell=True, check=True)
return outfile
def parse_hmmsearch_tbl(tbl_file: str) -> list[dict]:
"""Parse hmmsearch --tblout output."""
results = []
with open(tbl_file) as f:
for line in f:
if line.startswith("#"):
continue
parts = line.split()
if len(parts) < 10:
continue
results.append({
"target": parts[0], "query": parts[2],
"e_value": float(parts[4]), "score": float(parts[5])
})
return results
7. Visualization
7a. ETE3 tree plot (recommended)
from ete3 import Tree, TreeStyle, NodeStyle, TextFace, faces
import matplotlib.pyplot as plt
# Color palette (colorblind-friendly)
LINEAGE_COLORS = {
"DRT3": "#E69F00", # orange
"Retroviral": "#56B4E9", # sky blue
"GroupII": "#009E73", # green
"Retron": "#F0E442", # yellow
"Telomerase": "#0072B2", # blue
"LINE": "#D55E00", # vermillion
"RdRp": "#CC79A7", # pink (outgroup)
"default": "#999999" # gray
}
def plot_tree_ete3(treefile: str, output_png: str,
lineage_map: dict[str, str], # {taxon_name: lineage_label}
support_threshold: float = 70.0,
title: str = "") -> None:
"""
Plot a phylogenetic tree with colored tips and bootstrap support labels.
lineage_map: maps each leaf name to a lineage category for coloring.
"""
t = Tree(treefile)
ts = TreeStyle()
ts.show_leaf_name = False
ts.show_branch_support = False
ts.mode = "r" # rectangular
ts.scale = 200
if title:
ts.title.add_face(TextFace(title, fsize=14), column=0)
for node in t.traverse():
ns = NodeStyle()
ns["size"] = 0
if node.is_leaf():
lineage = lineage_map.get(node.name, "default")
color = LINEAGE_COLORS.get(lineage, "#999999")
ns["fgcolor"] = color
ns["size"] = 6
label = TextFace(f" {node.name}", fsize=9, fgcolor=color)
node.add_face(label, column=0, position="branch-right")
else:
# Show support on internal nodes if above threshold
try:
support = float(node.support)
if support >= support_threshold:
sf = TextFace(f"{support:.0f}", fsize=7, fgcolor="#333333")
node.add_face(sf, column=0, position="branch-top")
except (ValueError, TypeError):
pass
node.set_style(ns)
t.render(output_png, tree_style=ts, dpi=150, w=800)
7b. matplotlib fallback (no ETE3)
import matplotlib.pyplot as plt
import matplotlib.patches as mpatches
from Bio import Phylo
import io
def plot_tree_matplotlib(treefile: str, output_png: str,
lineage_map: dict[str, str],
figsize: tuple = (10, 12),
title: str = "") -> None:
"""Matplotlib-based tree plot using Bio.Phylo."""
tree = Phylo.read(treefile, "newick")
tree.ladderize()
fig, ax = plt.subplots(figsize=figsize)
Phylo.draw(tree, axes=ax, do_show=False,
label_func=lambda x: x.name if x.is_terminal() else "")
# Color leaf labels by lineage
for text in ax.texts:
name = text.get_text().strip()
lineage = lineage_map.get(name, "default")
color = LINEAGE_COLORS.get(lineage, "#999999")
text.set_color(color)
if title:
ax.set_title(title, fontsize=13)
ax.axis("off")
# Legend
legend_patches = [mpatches.Patch(color=v, label=k)
for k, v in LINEAGE_COLORS.items() if k != "default"]
ax.legend(handles=legend_patches, loc="lower left", fontsize=8)
plt.tight_layout()
plt.savefig(output_png, dpi=150, bbox_inches="tight")
plt.close()
8. Output Deliverables
Save all outputs to /mnt/results/ with a consistent prefix (e.g., drt3_rt):
| File | Description |
|---|---|
<prefix>_phylogeny.png |
Annotated tree figure (color-coded by lineage, bootstrap labels) |
<prefix>_phylogeny.treefile |
Newick tree with SH-aLRT/UFBoot support values |
<prefix>_alignment.fasta |
Trimmed multiple sequence alignment |
<prefix>_sequences.fasta |
Full input sequences before trimming |
report_<prefix>_phylogeny.md |
Methods, results, caveats, key findings |
Report template
# Phylogenetic Analysis: <title>
**Date:** YYYY-MM-DD
**Sequences:** N taxa, source (UniProt/NCBI/PDB)
**Domain:** <domain name, e.g., RT palm domain, ~200 aa>
## Methods
- Alignment: MAFFT <algorithm>, <N_cols_before> → <N_cols_after> columns after trimming
- Model: <best-fit model from ModelFinder>
- Tree: IQ-TREE2, <bootstrap type> (N replicates)
- Rooting: <outgroup name or midpoint>
- Ancestral reconstruction: <tool and method if used>
## Results
- Key topology: <describe placement of focal taxa>
- Support: <key node support values>
- Branch lengths: <notable long/short branches>
- Motifs found: <YXDD positions, etc.>
## Caveats
- <List any LBA risks, low support nodes, alignment issues>
9. Known Caveats and Pitfalls
Long-branch attraction (LBA)
- Symptom: A fast-evolving taxon (long branch) is pulled toward the outgroup or another long branch, regardless of true relationship.
- Detection: Branch length >3× mean branch length; check with
flag_long_branches(). - Mitigation: Use LG+I+G4 or WAG+I+G4 (rate heterogeneity helps); try removing the long-branch taxon and check if topology changes; use Bayesian methods (BEAST2) as a cross-check.
- Example: Drt3b (2.27 subs/site) in the DRT3 RT phylogeny — LBA risk flagged.
Alignment saturation
- Symptom: Very short alignment after trimming (<50 columns), or pi_fraction < 0.3.
- Detection: Check
alignment_quality()output. - Mitigation: Expand domain window (increase padding in
trim_sequence_to_domain); use less aggressive trimming (max_gap_frac=0.7 instead of 0.5).
UniProt accession mismatches
- Symptom: Fetched sequence has wrong length or organism.
- Detection: Always verify
len(seq)and organism name after fetch. - Mitigation: Cross-check with NCBI; use
fetch_ncbi_protein()as fallback.
AlphaFold coverage gaps for viral polyproteins
- Symptom: Many viral RT/RdRp sequences have no AlphaFold entry.
- Mitigation: Use PDB structures directly; or use cellular homologs (e.g., Arabidopsis RdRp6 Q9SG02 as RdRp representative).
IQ-TREE2 UFBoot inflation for small datasets
- Symptom: UFBoot values appear high (>90%) for a dataset with <50 taxa.
- Mitigation: Use
-b 100(standard bootstrap) instead of-bb 1000for datasets <50 taxa.
Outgroup too distant
- Symptom: Outgroup branch is extremely long; ingroup topology is unstable.
- Mitigation: Use a closer outgroup; or use midpoint rooting.
10. Worked Example: DRT3 RT Phylogeny
This skill was developed and validated on the DRT3 RT phylogeny analysis (Pedro Torres, LMDM/UFRJ, 2026).
Input: 20 sequences spanning 9 RT lineages (DRT3a/b, DRT9, retroviral, group II intron, retron, telomerase, LINE-1, pararetrovirus, RdRp outgroup).
Domain: RT palm domain (~200 aa, extracted around YXDD motif ± 100 aa).
Alignment: MAFFT L-INS-i → 821 columns → 193 after 50% gap trimming. 174/193 parsimony-informative sites (90.2%).
Tree: IQ-TREE2, LG+I+G4 (ModelFinder), 1000 UFBoot + SH-aLRT, rooted on RdRp (Q9SG02).
Key result: DRT3 falls within the UG/Abi clade (SH-aLRT=89.1, UFBoot=73 for broader clade). Group II intron RTs are more basal (older). DRT3-specific nodes have UFBoot=39–44 (not resolved). Drt3b has the longest branch (2.27 subs/site) — LBA risk flagged.
Motifs found:
- Drt3a: YVDD at position 194 (motif C), LPPG at position 219 (motif B)
- Drt3b: YVDD at position 288 (motif C); motif B absent
Output files: drt3_rt_phylogeny.png, drt3_rt_phylogeny.treefile, drt3_rt_alignment.fasta, drt3_rt_sequences.fasta
Code preview
No Python/R preview files were found.
Companion files
| Type | Path | Bytes |
|---|---|---|
| Markdown | SKILL.md | 24,764 |
| JSON | skill.meta.json | 855 |