DE — Auto Method Selection
Unsure which DE method? It picks based on your data.
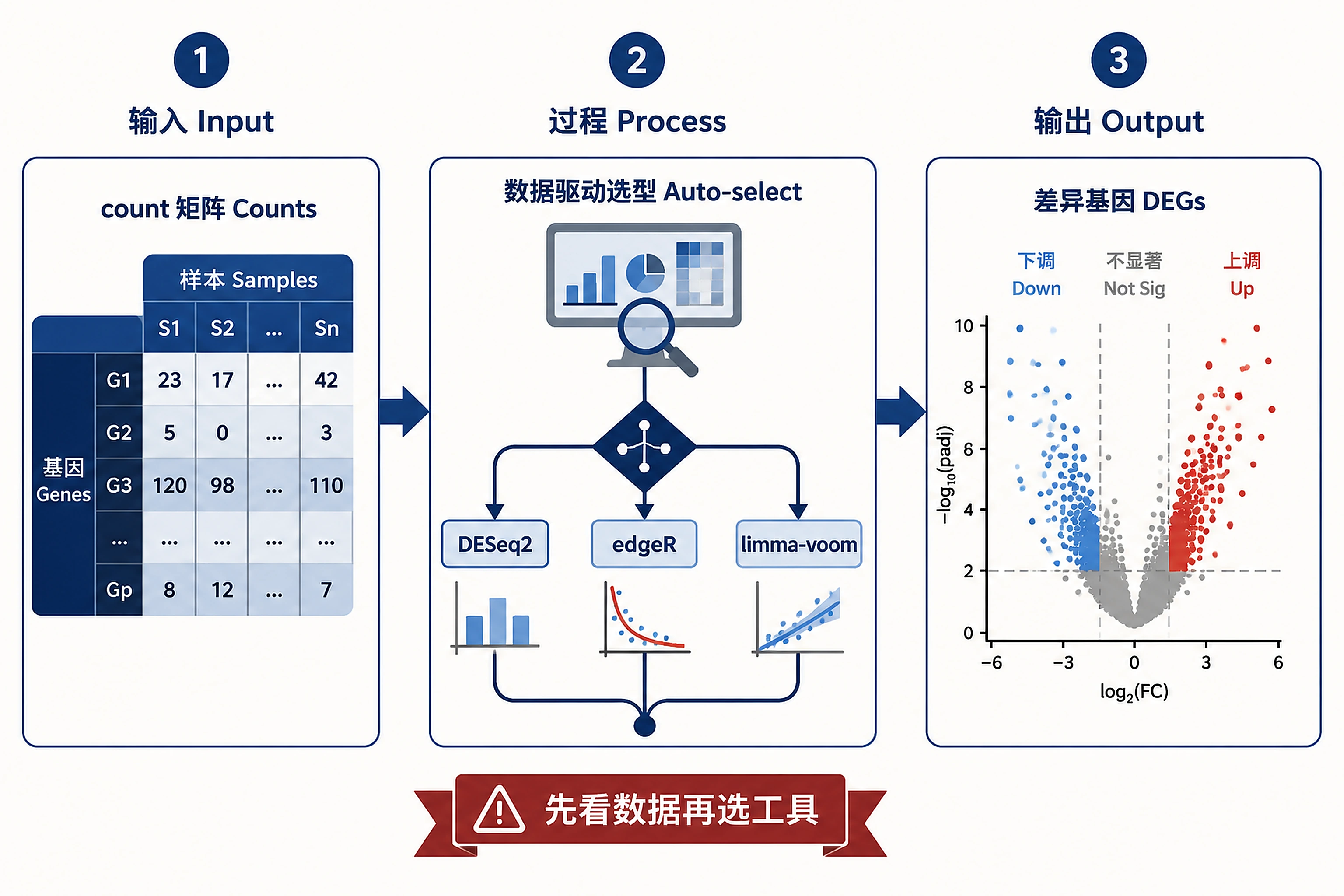
Overview
Problem. Data-driven choice among the three mainstream DE methods.
Learning goals
- Small n→edgeR, normalised/large→limma-voom, medium-large→DESeq2
- Inspect the data before choosing a tool
Figures
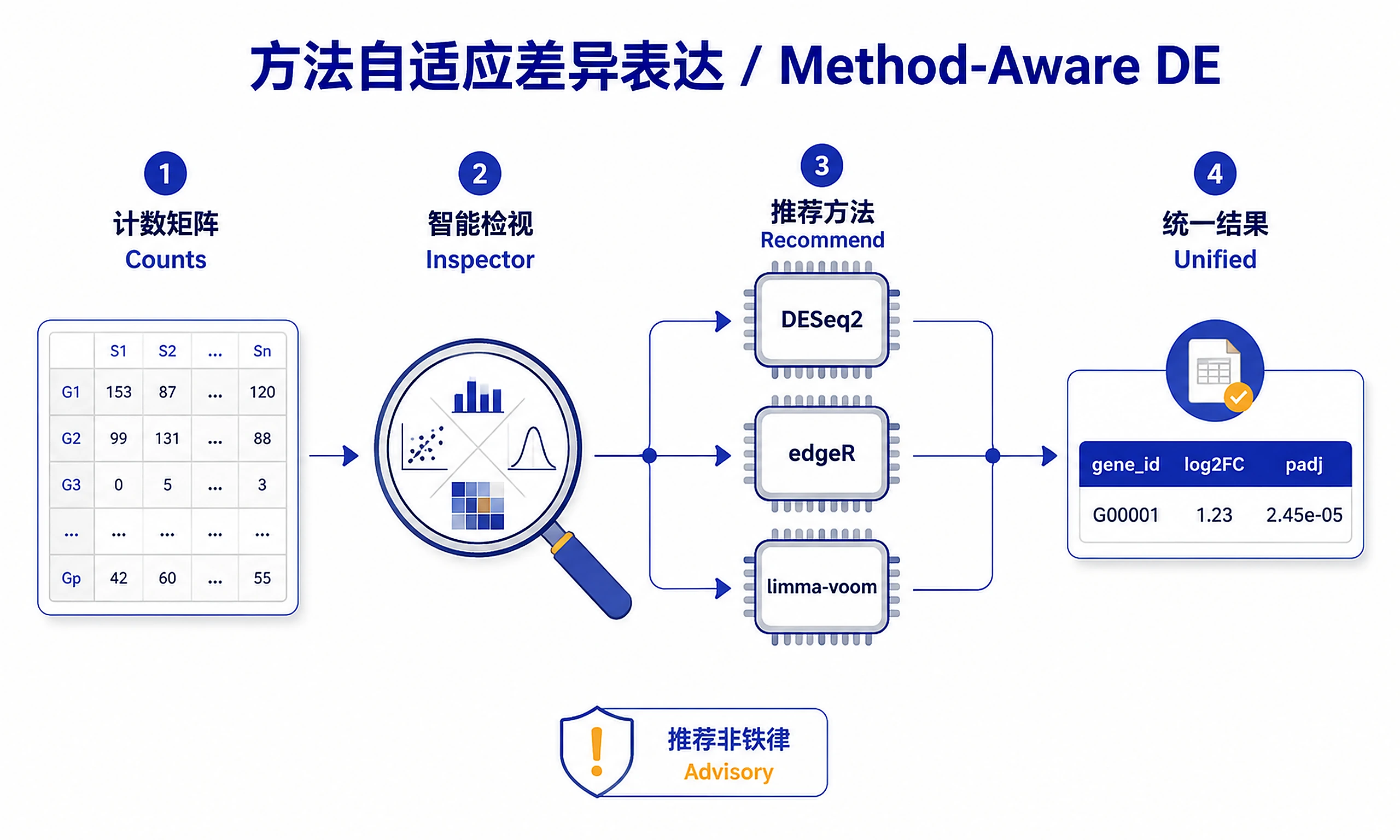
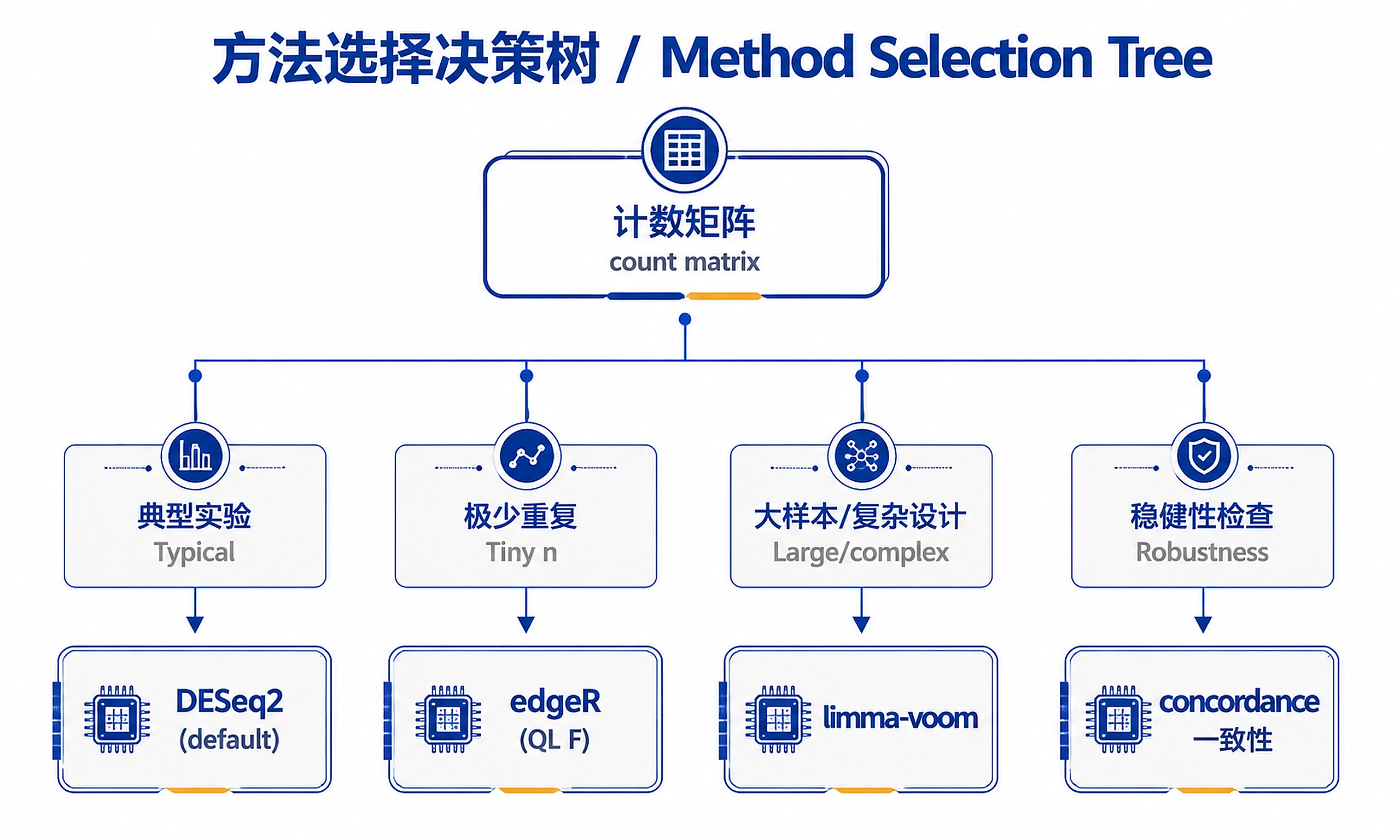

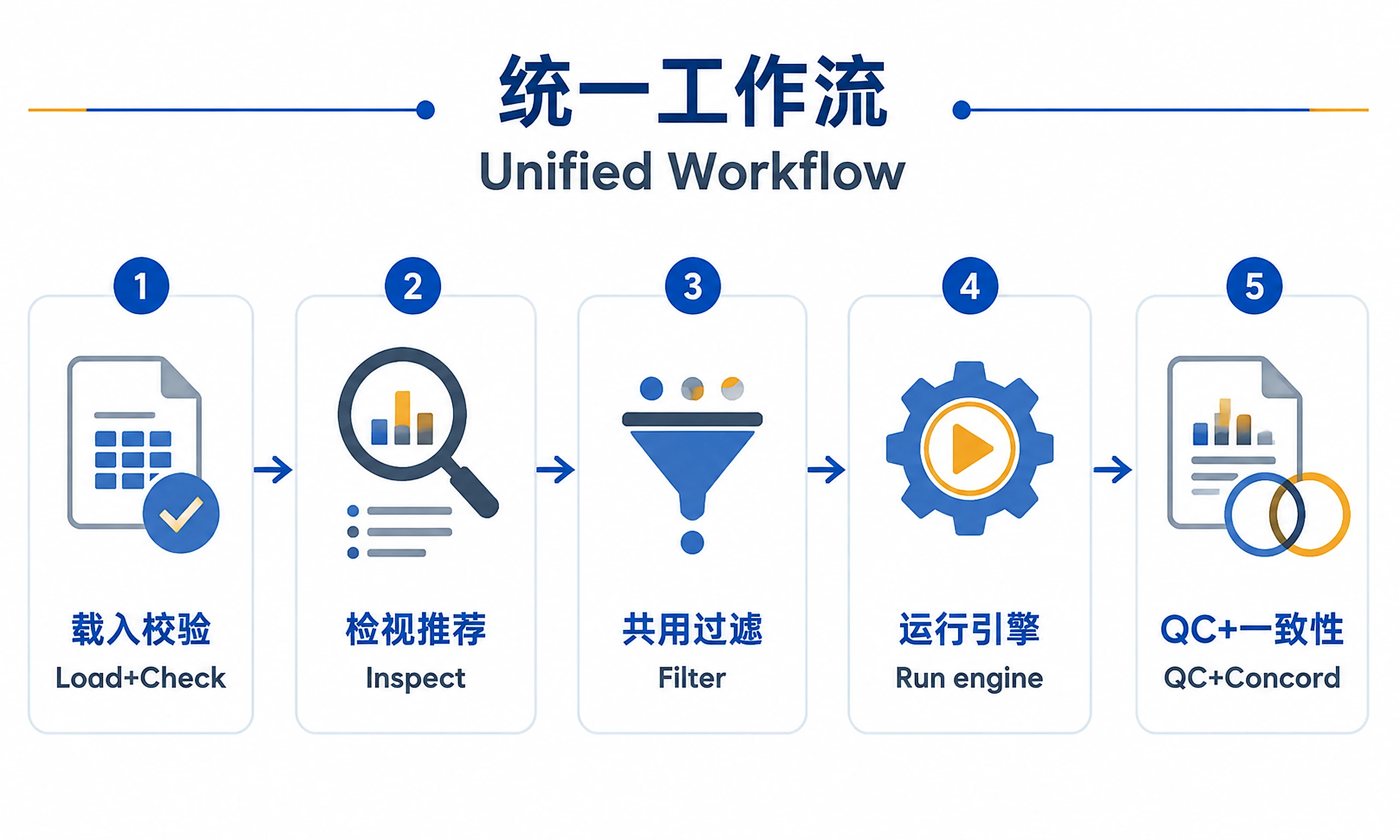

Tutorial
A method-aware differential expression skill for bulk RNA-seq count data. It does not lock you into one engine. Instead it looks at your data, recommends DESeq2, edgeR, or limma-voom with a plain-language rationale, lets you confirm or override, then runs a single unified workflow and (optionally) cross-checks gene calls across methods.
Reach for this whenever someone has a bulk RNA-seq count matrix and wants differentially expressed genes — especially when the question is "which method should I use?"
When to Use This Skill
Use it when the user has a bulk RNA-seq count matrix and wants differentially expressed genes, and any of the following is true:
- They don't specify a method, or explicitly aren't sure whether to use DESeq2, edgeR, or limma-voom.
- The experiment has small n, large n, or a non-trivial design (batch, paired, multi-group).
- They want a robustness/sensitivity check across methods (concordance).
- They uploaded
quant.sf(Salmon), Kallisto, featureCounts, or aSummarizedExperiment.
Not for: single-cell RNA-seq (use a scRNA-seq skill), normalized-only data where raw counts are unavailable (handled only as a flagged last-resort fallback — see Caveats), or upstream alignment/quantification (counts are the entry point).
Inputs
| Input | Format | Notes |
|---|---|---|
| Count matrix | genes x samples, raw integer counts | CSV/TSV/RDS, tximport (Salmon/Kallisto), featureCounts, or SummarizedExperiment. Not TPM/FPKM/log-normalized. |
| Sample metadata | data frame | Rownames (or a sample-id column) match count columns. Must contain a grouping column (default name condition). Optional: batch, individual/subject, other covariates. |
| User options (optional) | — | Method override, reference level, design formula, which contrast/coefficient to test, significance thresholds, whether to run concordance. |
The mechanics of loading, validating, and orienting the matrix live in
scripts/load_data.R — use it rather than reimplementing ingestion.
Outputs (saved to the results directory)
de_results_<method>.csv— full standardized results table.de_significant_<method>.csv— significant genes (default padj < 0.05) plus the |log2FC| ≥ 1 subset.- QC plots (PNG and SVG): PCA, MA, volcano, and the method-appropriate mean-variance / dispersion plot.
normalized_counts.csvand the fitted model object (.rds).- On request:
concordance_table.csv+consensus_degs.csv. run_log.txt— method chosen and why, design formula, contrast tested, filter summary, thresholds, full-rank check result, per-enginebaseMean_equiv_scale, and package versions.
Standardized results schema (identical across all three engines)
gene_id, baseMean_equiv, log2FoldChange, pvalue, padj, method
This common schema is what makes downstream tooling and concordance method-agnostic. Two columns require care:
baseMean_equivis a method-native expression-strength metric and is NOT comparable across methods. DESeq2 reports it on the linear count scale (mean normalized count); edgeR (logCPM) and limma (AveExpr) report it on the log2 scale. The same gene's value can differ by orders of magnitude purely because of which engine ran. It is fine for an engine's own MA plot and within-method ranking, but never use it in cross-method logic. Seereferences/comparison-and-caveats.md.- There is deliberately no merged
statcolumn. Wald z (DESeq2), QL F (edgeR), and moderated t (limma) are not comparable, so each engine keeps its native statistic only in its own raw output (labeled with astat_type); it is never cross-merged.
Workflow
The hard mechanics are in the scripts; your job is to drive them in the right order and
explain the method choice. Run from the skill directory so relative scripts/... paths
resolve.
Step 0 — Clarify inputs (ask first).
Step 1 — Load and validate.
source("scripts/load_data.R")
loaded <- load_de_data(counts = "<path-or-matrix>", metadata = "<path-or-df>",
condition_col = "condition")
This checks sample-ID concordance, matrix orientation, and integer-ness, and returns a
clean counts matrix + coldata data frame.
Step 2 — Inspect and recommend (advisory).
source("scripts/inspect_and_recommend.R")
rec <- inspect_and_recommend(loaded$counts, loaded$coldata,
condition_col = "condition",
design = ~ condition) # or your covariate formula
print(rec)
This reports replicates per group, integer-ness, library-size spread, PCA clustering by condition/batch, and a programmatic full-rank/confounding check (model-matrix rank vs its number of columns). It returns a recommended method and a proposed design formula.
Translate rec into a short prose recommendation for the user — prefer the recommended
method, explain why, name the alternatives, and ask them to confirm or override. The
heuristics are advisory, not hard rules:
- Default to DESeq2 for typical experiments — robust and widely validated.
- Lean toward edgeR when replicates per group are very small — its quasi-likelihood F-test handles tiny-n dispersion estimation well.
- Lean toward limma-voom when sample size is large or the design is complex/multi-factor — it is fast, well-calibrated, and flexible with model formulas.
- If more than one method is reasonable, recommend the default and offer the concordance check rather than forcing a single choice.
If inspect_and_recommend reports the design is not full rank (e.g. batch perfectly
aliased with condition), stop and resolve the confounding with the user before running —
do not silently proceed.
Step 3 — Confirm.
Step 4 — Shared pre-filter (always, before any engine).
source("scripts/filter_counts.R")
filt <- filter_counts(loaded$counts, loaded$coldata, condition_col = "condition")
This applies one common filterByExpr() low-expression filter on the raw counts. The
same filtered gene set is reused by whichever engine(s) run — this is what makes a later
concordance comparison reflect method differences rather than filtering differences.
Step 5 — Run the chosen engine
source("scripts/run_deseq2.R") # or run_edger.R / run_limma_voom.R
fit <- run_deseq2(filt$counts, filt$coldata,
design = ~ condition,
contrast = c("condition", "treated", "control"), # or coef name
ref_level = "control")
res <- fit$results # standardized data frame
For DESeq2, apeglm LFC shrinkage is applied for the engine's own ranking/visualization;
the standardized log2FoldChange reported for cross-method use is the unshrunk value.
Step 6 — QC plots and export.
source("scripts/qc_plots.R"); run_all_qc(fit, output_dir = "results")
source("scripts/export_results.R"); export_de(fit, output_dir = "results")
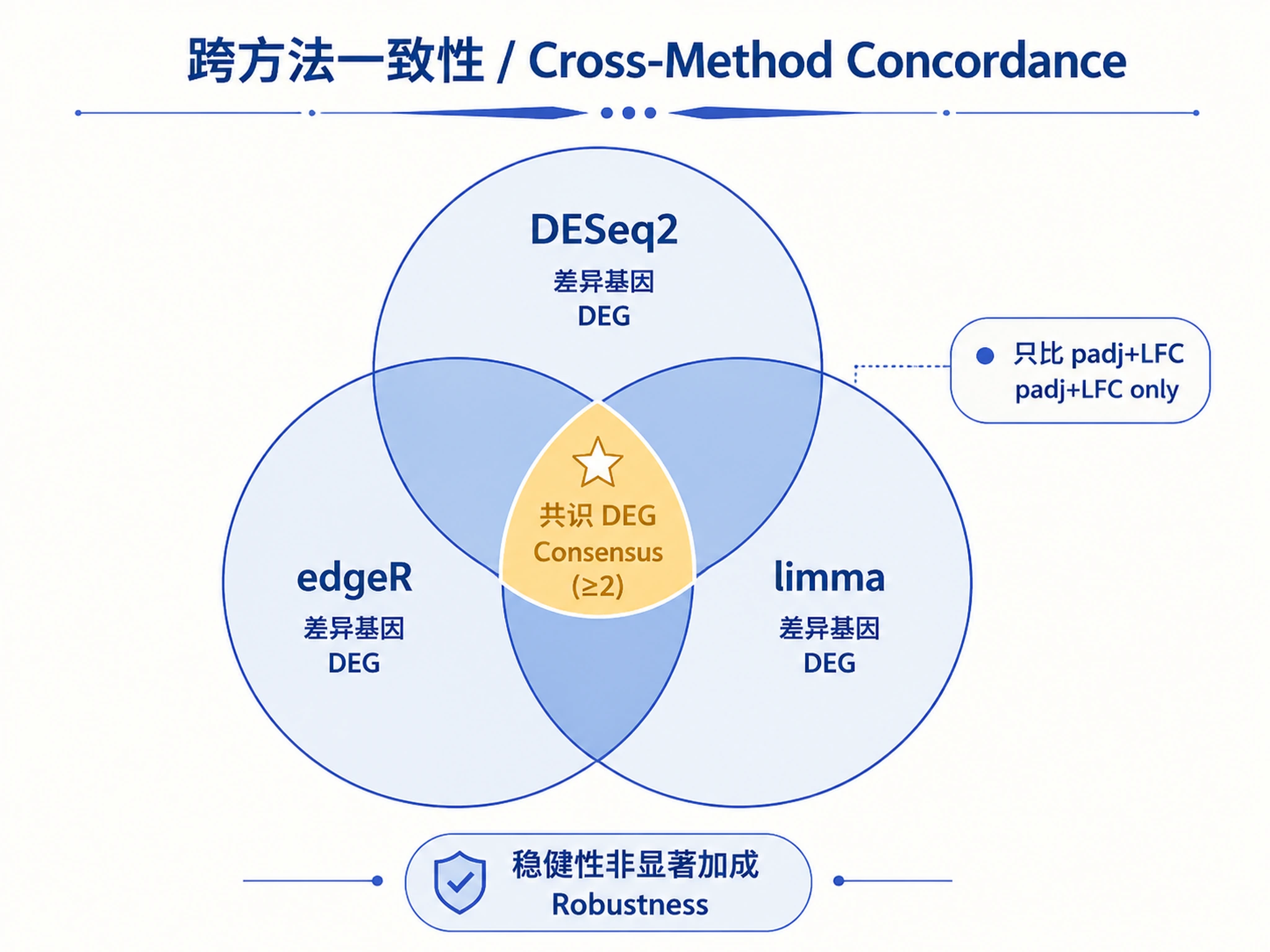
Step 7 — (On request) cross-method concordance.
source("scripts/concordance.R")
concordance(list(deseq2 = res_deseq2, edger = res_edger, limma = res_limma),
padj_cutoff = 0.05, output_dir = "results")
This produces an overlap-count table and a consensus DEG list (genes significant in ≥2
methods). It compares using only padj and unshrunk log2FoldChange — never
baseMean_equiv. Concordance is a robustness check, not a significance booster.
Step 8 — Report.
Method Selection at a Glance (advisory)
| Situation | Lean toward | Why |
|---|---|---|
| Typical experiment, raw counts, simple design | DESeq2 (default) | Robust, validated, good default shrinkage |
| Very small replicates per group | edgeR (QL F-test) | Quasi-likelihood handles tiny-n dispersion |
| Large n and/or complex multi-factor design | limma-voom | Fast, well-calibrated, flexible formulas |
| User wants a robustness check | run ≥2 + concordance | Reports gene-call overlap across methods |
These are starting points to explain and confirm with the user, not thresholds to apply
mechanically. Full narrative: references/method-selection-guide.md.
Scientific Caveats
- Use adjusted p-values (BH-FDR
padj), never raw p-values, to call significance — thousands of genes are tested simultaneously. - Count-based methods (DESeq2, edgeR) and limma-voom all require raw integer counts,
never TPM/FPKM. If only normalized values exist,
log2(TPM)→ limma-trend is a documented last-resort fallback with strong caveats: TPM's transcript-length normalization violates the logCPM mean–variance assumption, so results are approximate and must be flagged as such. - The design must be full rank / not confounded. This is checked programmatically in
inspect_and_recommend.R; resolve any confounding before running. baseMean_equivis not comparable across methods (see schema note above).- Cross-method LFCs are only compared on unshrunk terms in concordance output.
Self-Test / Eval
scripts/selftest.R runs all three engines on a bundled example
dataset (pasilla) and asserts: every engine emits the standardized schema; significance
uses padj; the shared filter yields one common gene universe; the recommender behaves
sensibly across n; the full-rank check flags a confounded design; concordance uses only
padj/LFC (never baseMean_equiv); and a non-integer matrix triggers the raw-counts
guardrail. Run it after installing the engine packages to validate the skill end-to-end.
References
references/method-selection-guide.md— when and why to choose each engine (narrative).references/comparison-and-caveats.md— assumptions, FDR, design/contrast handling, LFC- and baseMean-comparability.references/troubleshooting.md— common errors across all three engines.
Code preview
scripts/concordance.R
# =============================================================================
# concordance.R — on-demand cross-method DEG concordance (TABLE ONLY)
#
# Compares significant-gene calls across 2+ engines that were run on the SAME
# shared-filtered gene universe (see filter_counts.R). This is a ROBUSTNESS check,
# NOT a way to gain significance.
#
# Comparability rules (enforced here):
# - Significance is decided ONLY by `padj` (BH-FDR) at `padj_cutoff`
# (optionally combined with |log2FoldChange| >= lfc_cutoff if lfc_cutoff > 0).
# - The consensus list reports the UNSHRUNK `log2FoldChange` carried in each
# engine's standardized table (the engines already report unshrunk LFC).
# - `baseMean_equiv` is NEVER used here (its scale differs by engine).
#
# Inputs:
# results_list : named list of standardized data.frames from run_*() ($results),
# e.g. list(DESeq2 = res1, edgeR = res2, `limma-voom` = res3)
# padj_cutoff : significance threshold (default 0.05)
# lfc_cutoff : optional |log2FC| filter applied to significance (default 0 = none)
# min_methods : a gene is "consensus" if significant in >= this many methods (default 2)
#
# Writes (to output_dir):
# concordance_table.csv : per-method DEG counts + pairwise overlaps + Jaccard
# consensus_degs.csv : genes significant in >= min_methods, with each method's
# unshrunk log2FC and padj, and n_methods_significant
# Returns these as a list (invisibly).
# =============================================================================
concordance <- function(results_list, padj_cutoff = 0.05, lfc_cutoff = 0,
min_methods = 2L, output_dir = "results") {
stopifnot(length(results_list) >= 2L)
dir.create(output_dir, showWarnings = FALSE, recursive = TRUE)
if (is.null(names(results_list)) || any(names(results_list) == "")) {
names(results_list) <- vapply(results_list, function(r) unique(r$method)[1], character(1))
}
methods <- names(results_list)
# --- guardrail: refuse to touch baseMean_equiv in any cross-method computation ---
# (defensive: ensure callers didn't pass a column we must not compare)
sig_set <- function(df) {
keep <- !is.na(df$padj) & df$padj < padj_cutoff
if (lfc_cutoff > 0) keep <- keep & is.finite(df$log2FoldChange) &
abs(df$log2FoldChange) >= lfc_cutoff
df$gene_id[keep]
}
sig_lists <- lapply(results_list, sig_set)
names(sig_lists) <- methods
# universe = union of all gene_ids tested (should be identical if shared-filtered)
universes <- lapply(results_list, function(d) d$gene_id)
shared_ok <- length(unique(lapply(universes, function(u) sort(unique(u))))) == 1L
# ---------- summary / overlap table ----------
rows <- list()
for (mi in methods) {
rows[[length(rows) + 1]] <- data.frame(
comparison = mi, type = "n_significant",
value = length(sig_lists[[mi]]), stringsAsFactors = FALSE)
}
# all pairwise overlaps + Jaccard
if (length(methods) >= 2L) {
cb <- utils::combn(methods, 2L)
for (k in seq_len(ncol(cb))) {
a <- cb[1, k]; b <- cb[2, k]
inter <- length(intersect(sig_lists[[a]], sig_lists[[b]]))
uni <- length(union(sig_lists[[a]], sig_lists[[b]]))
rows[[length(rows) + 1]] <- data.frame(
comparison = paste(a, b, sep = " & "), type = "overlap",
value = inter, stringsAsFactors = FALSE)
rows[[length(rows) + 1]] <- data.frame(
comparison = paste(a, b, sep = " & "), type = "jaccard",
value = if (uni > 0) round(inter / uni, 4) else NA_real_,
stringsAsFactors = FALSE)
}
}
# genes significant in ALL methods
all_inter <- Reduce(intersect, sig_lists)
rows[[length(rows) + 1]] <- data.frame(
comparison = paste(methods, collapse = " & "), type = "overlap_all",
value = length(all_inter), stringsAsFactors = FALSE)scripts/export_results.R
# =============================================================================
# export_results.R — standardized exports + run log
#
# Works on the `fit` object returned by any run_*() engine. All engines return
# the same structure:
# fit$results : standardized data.frame
# (gene_id, baseMean_equiv, log2FoldChange, pvalue, padj, method)
# fit$method : "DESeq2" | "edgeR" | "limma-voom" | "limma-trend"
# fit$baseMean_scale : "linear" | "log2" (scale of baseMean_equiv for THIS engine)
# fit$stat_type : native statistic label (Wald z / QL F / moderated t)
# fit$normalized : normalized expression matrix (for export)
# fit$object : the underlying fitted object (for saveRDS)
# fit$meta : list with design, contrast, ref_level, filter_summary,
# full_rank, package_version, thresholds, ...
#
# Writes (to output_dir):
# de_results_<method>.csv
# de_significant_<method>.csv (padj < cutoff)
# de_significant_fc_<method>.csv (padj < cutoff & |log2FC| >= lfc_cutoff)
# normalized_counts_<method>.csv
# de_fit_<method>.rds
# run_log.txt (appended; one block per engine run)
# =============================================================================
# Null/NA-coalescing helper (defined first so it is available throughout).
`%||%` <- function(a, b) if (is.null(a) || length(a) == 0L ||
(length(a) == 1L && is.na(a))) b else a
# saveRDS to S3-backed mounts (/mnt/results, /mnt/shared-workspace) fails or
# yields 0-byte files because .rds uses random-access writes. Write to local
# scratch first, then shell-copy into place.
.safe_saveRDS <- function(object, path) {
on_s3 <- grepl("^/mnt/(results|shared-workspace)", normalizePath(dirname(path),
mustWork = FALSE))
if (on_s3) {
tmp <- file.path(tempdir(), basename(path))
saveRDS(object, tmp)
ok <- file.copy(tmp, path, overwrite = TRUE)
# R's file.copy can also 0-byte on FUSE; fall back to shell cp if so.
if (!isTRUE(ok) || file.info(path)$size %in% c(0, NA)) {
system2("cp", c(shQuote(tmp), shQuote(path)))
}
unlink(tmp)
} else {
saveRDS(object, path)
}
invisible(path)
}
export_de <- function(fit, output_dir = "results",
padj_cutoff = 0.05, lfc_cutoff = 1) {
dir.create(output_dir, showWarnings = FALSE, recursive = TRUE)
m <- fit$method
res <- fit$results
# ---- standardized full table ----
f_all <- file.path(output_dir, sprintf("de_results_%s.csv", m))
utils::write.csv(res, f_all, row.names = FALSE)
# ---- significant (padj only; the headline definition) ----
sig <- res[!is.na(res$padj) & res$padj < padj_cutoff, , drop = FALSE]
sig <- sig[order(sig$padj), , drop = FALSE]
f_sig <- file.path(output_dir, sprintf("de_significant_%s.csv", m))
utils::write.csv(sig, f_sig, row.names = FALSE)
# ---- significant with fold-change subset ----
sig_fc <- sig[abs(sig$log2FoldChange) >= lfc_cutoff, , drop = FALSE]
f_sigfc <- file.path(output_dir, sprintf("de_significant_fc_%s.csv", m))
utils::write.csv(sig_fc, f_sigfc, row.names = FALSE)
# ---- normalized counts ----
f_norm <- NA_character_
if (!is.null(fit$normalized)) {
f_norm <- file.path(output_dir, sprintf("normalized_counts_%s.csv", m))
utils::write.csv(as.data.frame(fit$normalized), f_norm)
}
# ---- fitted object ----
# saveRDS uses random-access writes, which break on S3-backed mounts; the
# helper writes to local scratch then copies into place.scripts/filter_counts.R
# =============================================================================
# filter_counts.R — shared low-expression pre-filter for ALL engines
#
# Applies ONE common filter (edgeR::filterByExpr) to the raw count matrix using
# the grouping variable, so the SAME filtered gene set is handed to whichever
# engine(s) run. This is essential for fair cross-method concordance: otherwise
# the overlap would partly measure differences in each engine's own internal
# filtering rather than differences in the statistical methods.
#
# Returns: list(counts = filtered matrix, coldata = unchanged, keep = logical,
# n_before, n_after, filter_summary = character)
# =============================================================================
source_local <- function(f) {
# source a sibling script regardless of caller's working dir, if available
here <- tryCatch(dirname(sys.frame(1)$ofile), error = function(e) NA)
cand <- if (!is.na(here)) file.path(here, f) else f
if (file.exists(cand)) source(cand)
}
filter_counts <- function(counts, coldata, condition_col = "condition",
group = NULL, design = NULL, ...) {
options(repos = c(CRAN = "https://cloud.r-project.org"))
if (!requireNamespace("edgeR", quietly = TRUE)) {
if (!requireNamespace("BiocManager", quietly = TRUE)) install.packages("BiocManager")
BiocManager::install("edgeR", update = FALSE, ask = FALSE)
}
counts <- as.matrix(counts)
storage.mode(counts) <- "double"
if (is.null(group)) {
if (!condition_col %in% colnames(coldata)) {
stop("filter_counts: grouping column '", condition_col, "' not in coldata.")
}
group <- factor(coldata[[condition_col]])
}
n_before <- nrow(counts)
# filterByExpr keeps genes with a worthwhile count in a minimum number of
# samples, sized to the smallest group. Use the design if supplied (better for
# multi-factor models); otherwise use the group factor.
keep <- if (!is.null(design)) {
mm <- stats::model.matrix(design, data = coldata)
edgeR::filterByExpr(counts, design = mm)
} else {
edgeR::filterByExpr(counts, group = group)
}
counts_f <- counts[keep, , drop = FALSE]
n_after <- nrow(counts_f)
summary_txt <- sprintf(
"Shared filterByExpr pre-filter: %d -> %d genes kept (%d removed); applied identically before all engines.",
n_before, n_after, n_before - n_after)
message(summary_txt)
list(
counts = counts_f,
coldata = coldata,
keep = keep,
n_before = n_before,
n_after = n_after,
filter_summary = summary_txt
)
}Companion files
| Type | Path | Bytes |
|---|---|---|
| Markdown | references/comparison-and-caveats.md | 5,386 |
| Markdown | references/method-selection-guide.md | 4,721 |
| Markdown | references/troubleshooting.md | 6,138 |
| R | scripts/concordance.R | 6,092 |
| R | scripts/export_results.R | 5,548 |
| R | scripts/filter_counts.R | 2,590 |
| R | scripts/inspect_and_recommend.R | 9,315 |
| R | scripts/load_data.R | 6,980 |
| R | scripts/qc_plots.R | 5,752 |
| R | scripts/run_deseq2.R | 4,938 |
| R | scripts/run_edger.R | 5,397 |
| R | scripts/run_limma_voom.R | 5,988 |
| R | scripts/selftest.R | 16,427 |
| Markdown | SKILL.md | 10,939 |
| JSON | skill.meta.json | 3,020 |