scRNA Disease Drug Discovery
Single-cell plus genetics for multi-omics target prioritisation.
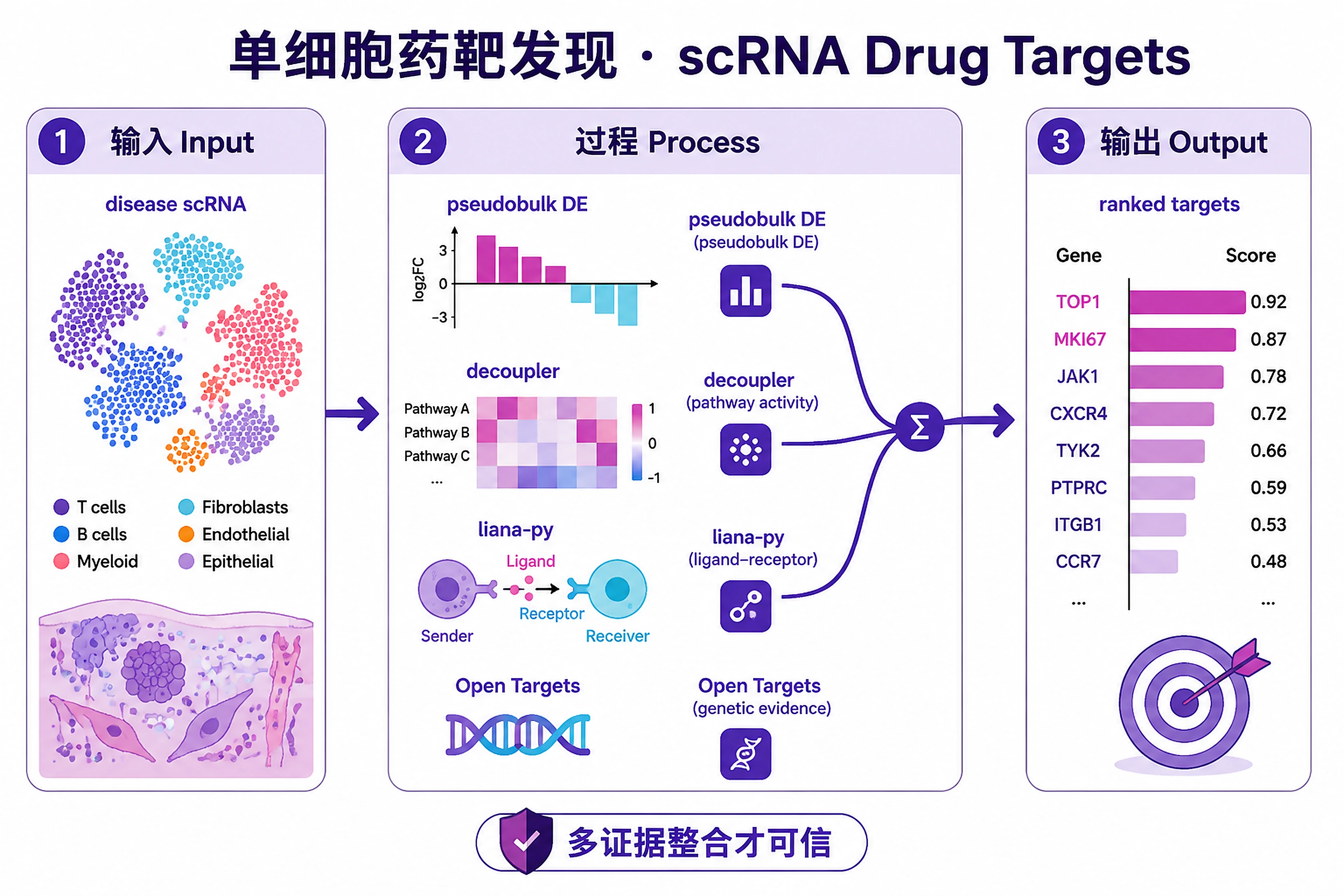
Overview
Problem. From disease scRNA to a ranked target list.
Learning goals
- How a real pipeline chains many skills
- Multiple evidence lines are what convince
Figures
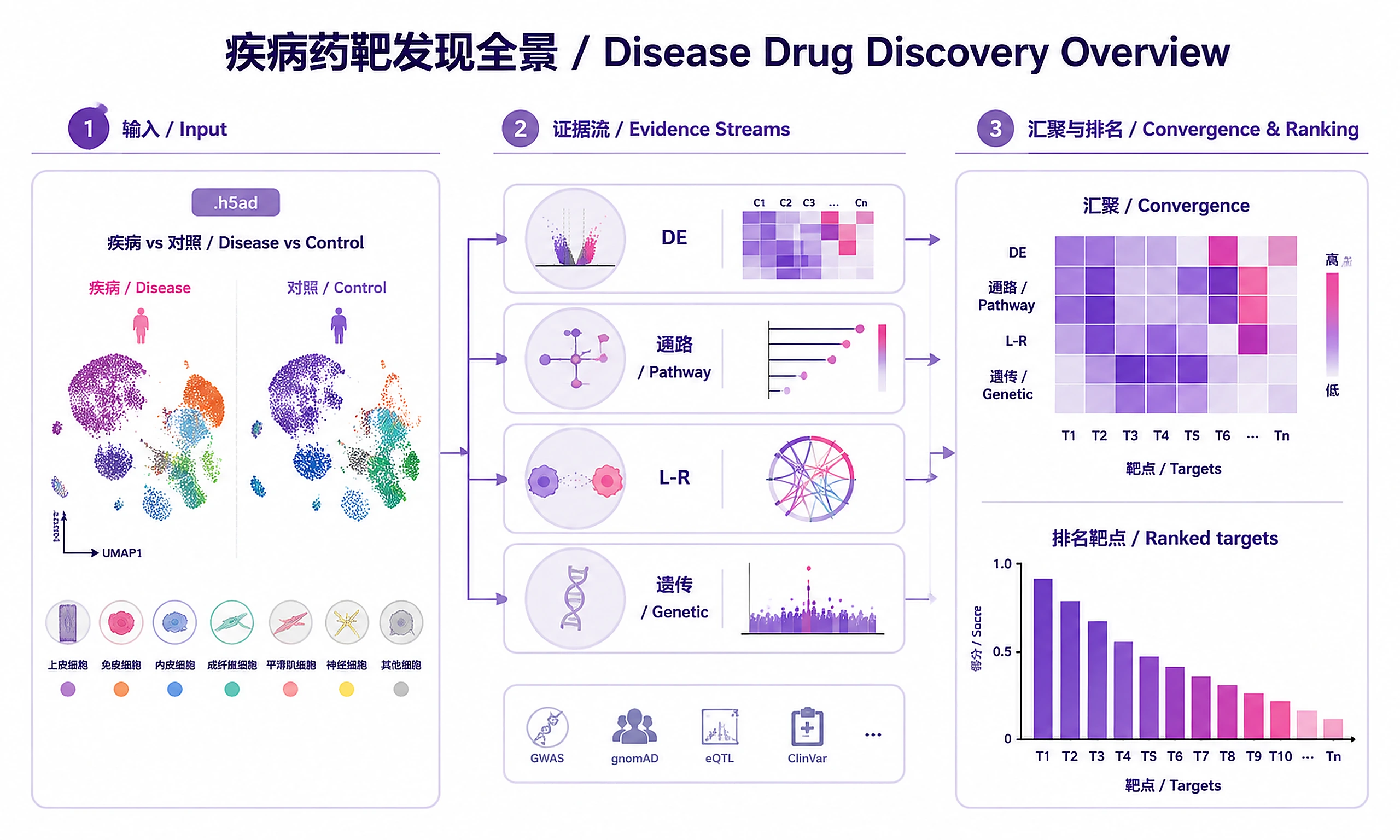
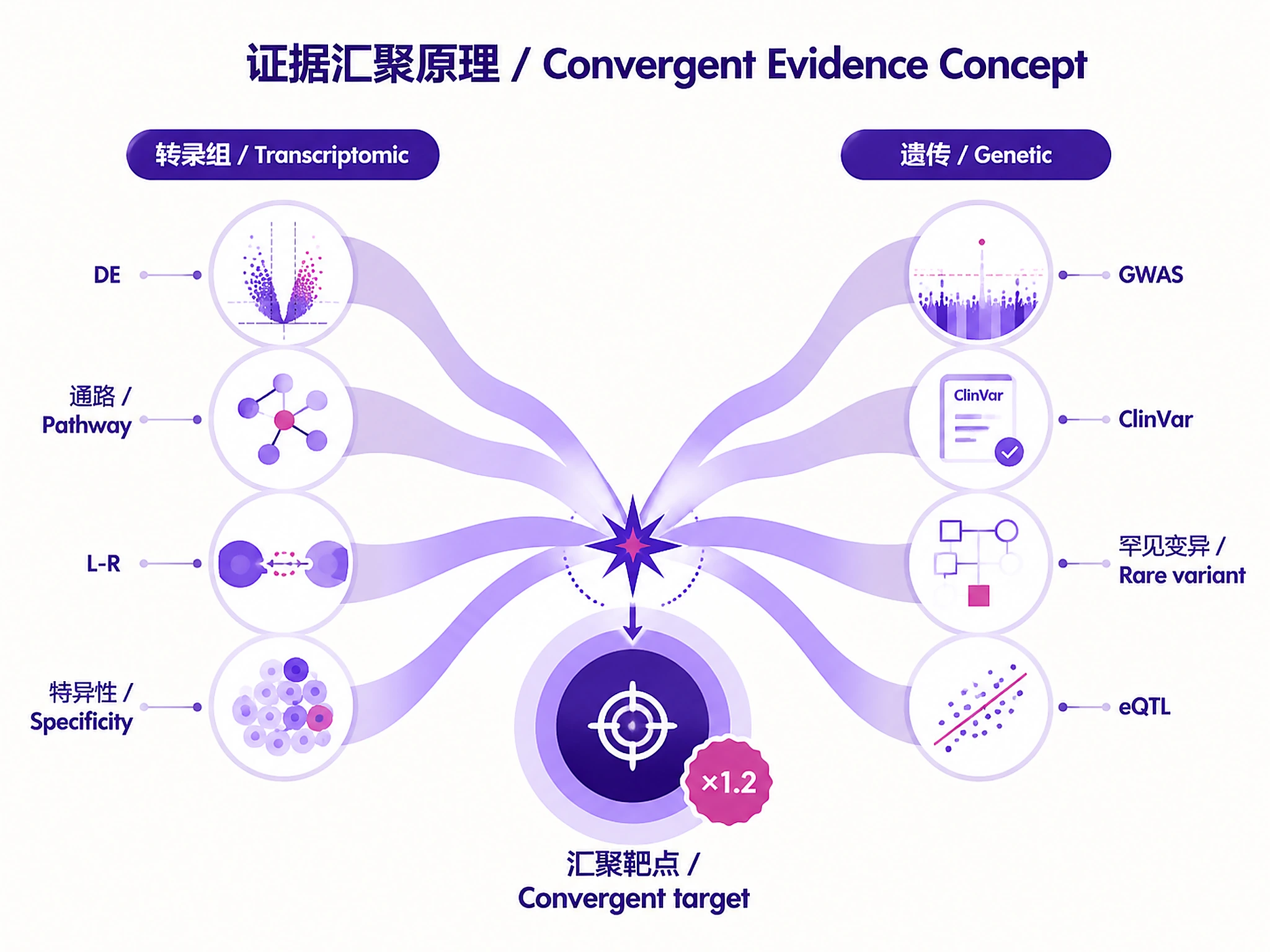
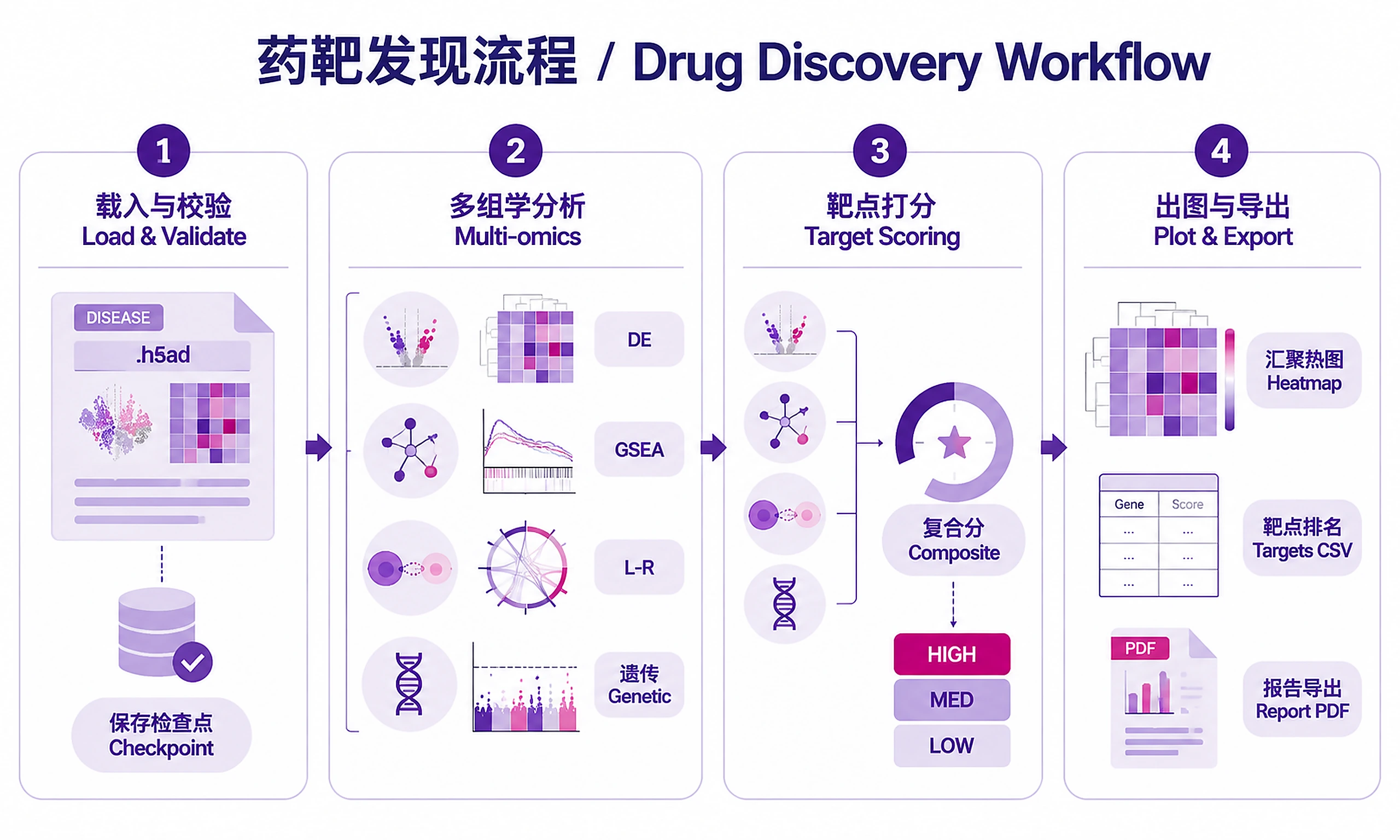
Tutorial
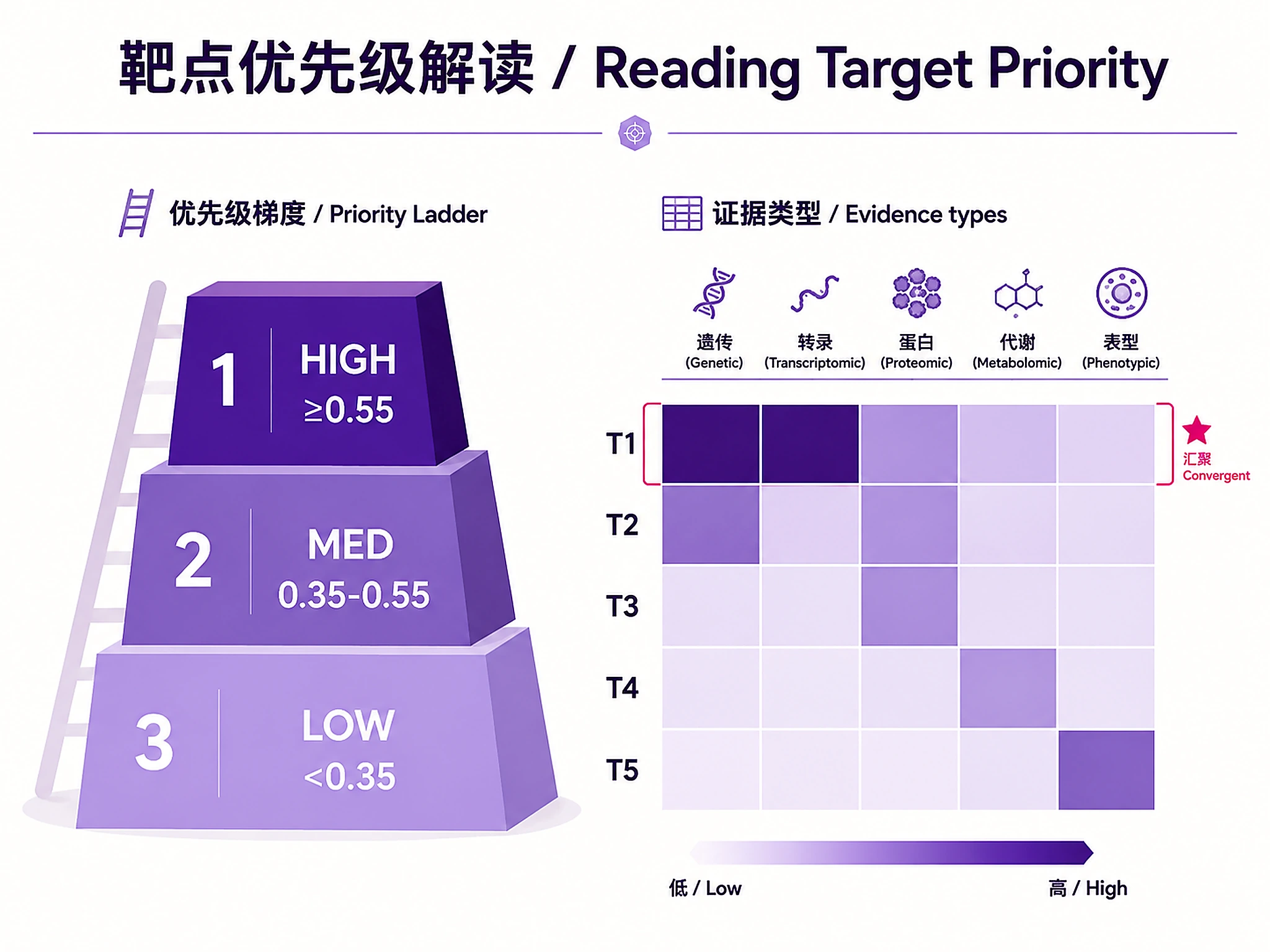
Identify and prioritize drug targets from disease scRNA-seq data by integrating transcriptomic evidence (differential expression, pathway enrichment, ligand-receptor interactions) with human genetic evidence (Open Targets GWAS/ClinVar, GeneBass rare variant burden, TWAS, eQTL). Targets with convergent multi-omics evidence receive the highest priority.
When to Use This Skill
- You have scRNA-seq data from a disease vs. control comparison and want therapeutic targets
- You want multi-omics target prioritization combining expression + genetic evidence
- You need cell-type-resolved drug discovery insights (which cell types drive disease?)
- You want to identify druggable targets with convergent transcriptomic and genetic support
Chains from scrnaseq-scanpy-core-analysis output (adata_processed.h5ad) or accepts raw data.
Don't use for:
- Cell type annotation only (use
scrnaseq-scanpy-core-analysis) - Genetic-only target discovery (use
genetic-target-hypothesis) - Gene regulatory networks (use
grn-pyscenic) - Spatial transcriptomics (use
spatial-transcriptomics)
Quick Start (Demo Data)
Test the full pipeline on SSc demo data (~20-40 min including API queries):
# Load demo dataset (GSE195452 SSc skin biopsies)
from load_data import load_demo_ssc_data
adata = load_demo_ssc_data()
# Run full pipeline
from pseudobulk_de import run_pseudobulk_de, get_disease_signature_genes
de_results = run_pseudobulk_de(adata, reference="Healthy", layer="counts")
from pathway_enrichment import run_pathway_analysis
pathway_results = run_pathway_analysis(adata, de_results)
from ligand_receptor import run_lr_analysis
lr_results = run_lr_analysis(adata)
sig_genes = get_disease_signature_genes(de_results)
candidate_genes = list(set(g for genes in sig_genes.values() for g in genes))[:200]
from genetic_evidence import collect_genetic_evidence
genetic_results = collect_genetic_evidence(candidate_genes, "systemic sclerosis")
from query_opentargets import query_target_annotations
ot_annotations = query_target_annotations(candidate_genes[:50])
from target_scoring import score_targets
scores = score_targets(de_results, pathway_results, lr_results, genetic_results, ot_annotations)
from generate_visualizations import generate_all_plots
generate_all_plots(adata, de_results, pathway_results, lr_results, genetic_results, scores)
from export_results import export_all
export_all(adata, de_results, pathway_results, lr_results, genetic_results, scores, ot_annotations)
What you get:
- Dataset: 277K cells from 165 SSc + healthy skin biopsies (subsampled for compute)
- Expected results: 100-700 DEGs across fibroblast subtypes, TGF-beta/ECM/IFN pathway enrichment, 15+ MEDIUM/HIGH priority targets with convergent genetic + transcriptomic evidence
- Outputs: ranked_targets.csv, 7+ PNG/SVG visualizations, per-cell-type DE CSVs, GSEA results, L-R interactions, genetic evidence summary, analysis_manifest.json
Installation
| Package | Version | License | Commercial Use | Installation |
|---|---|---|---|---|
| scanpy | >=1.9 | BSD-3-Clause | Permitted | pip install scanpy |
| anndata | >=0.8 | BSD-3-Clause | Permitted | pip install anndata |
| pydeseq2 | >=0.4 | MIT | Permitted | pip install pydeseq2 |
| decoupler | >=1.4 | MIT | Permitted | pip install decoupler |
| liana-py | >=0.1.7 | BSD-3-Clause | Permitted | pip install liana |
| gseapy | >=1.0 | BSD-3-Clause | Permitted | pip install gseapy |
| requests | >=2.28 | Apache-2.0 | Permitted | pip install requests |
| pyarrow | >=10.0 | Apache-2.0 | Permitted | pip install pyarrow |
| matplotlib | >=3.4 | PSF | Permitted | pip install matplotlib |
| seaborn | >=0.12 | BSD-3-Clause | Permitted | pip install seaborn |
| adjustText | >=0.8 | MIT | Permitted | pip install adjustText |
| numpy | >=1.20 | BSD-3-Clause | Permitted | pip install numpy |
| pandas | >=1.3 | BSD-3-Clause | Permitted | pip install pandas |
Install all: pip install scanpy anndata pydeseq2 decoupler liana gseapy requests pyarrow matplotlib seaborn adjustText
Optional (for preprocessing raw data): pip install scrublet celltypist harmonypy
Inputs
Required:
- Annotated AnnData (.h5ad) with cell type labels, condition labels, and sample/donor IDs
- Expected from
scrnaseq-scanpy-core-analysisor equivalent pipeline - Minimum: 3 cell types, >=10 cells per type, 2 conditions (disease + control)
OR:
- Raw/filtered count matrix (10X, H5, CSV) -- the skill will preprocess internally
Required metadata columns:
cell_type(or user-specified) -- cell type annotationscondition(or user-specified) -- disease status (e.g., "SSc", "Healthy")sample_id(or user-specified) -- biological replicate IDs for pseudobulk DE
Optional:
- Disease name for genetic evidence matching (default: inferred from condition labels)
- Species: Human (default) or Mouse
Outputs
Ranked target list:
ranked_targets.csv-- All scored targets with evidence columns: DE score, pathway score, L-R score, genetic score, druggability score, composite score, priority tier, convergence flagtarget_evidence_cards.json-- Per-target evidence summaries for report generation
Differential expression:
de_results_{celltype}.csv-- Per-cell-type DE results (gene, log2FC, padj, baseMean)de_summary.csv-- Cross-cell-type DEG summary
Pathway analysis:
pathway_enrichment_{celltype}.csv-- GSEA results per cell typepathway_activity_scores.csv-- decoupler pathway activity matrix
Ligand-receptor interactions:
lr_interactions.csv-- Significant L-R pairs with source/target cell types, methods, consensus ranklr_disease_specific.csv-- Disease-enriched interactions (disease vs control comparison)
Genetic evidence:
genetic_ot_disease_genetics.csv-- Open Targets GWAS/ClinVar/gene burden evidencegenetic_twas_associations.csv-- TWAS Atlas resultsgenetic_summary.csv-- Integrated genetic evidence per gene
Analysis objects:
adata_analyzed.h5ad-- AnnData with DE results, pathway scores in .uns- Load with:
import scanpy as sc; adata = sc.read_h5ad('adata_analyzed.h5ad') analysis_manifest.json-- Provenance: timestamp, parameters, step timing, output paths
Visualizations (PNG + SVG at 300 DPI):
- UMAP overview (cell type + condition)
- Volcano plots per cell type
- DEG heatmap across cell types (gene symbols, sorted by significance)
- Pathway activity heatmap (sorted by max |NES|, not alphabetical)
- L-R dot plot (disease-relevant interactions prioritized)
- Multi-omics convergence heatmap (targets x evidence types)
- Target ranking bar chart
Reports:
drug_discovery_report.pdf-- Comprehensive PDF with Methods, Results, Figures, Conclusions
PDF style rules:
- US Letter page size (8.5 x 11 in) -- always set explicitly
- No Unicode superscripts -- use
3.36e-06not Unicode chars - No half-empty pages -- group headings with content
- Figures >=80% page width
Decision Guide
| Decision | Quick Guide | Reference |
|---|---|---|
| Data type | 10X Chromium (default). Smart-seq2/plate: lower min_cells to 3-5 | references/workflow-details.md |
| Subsampling | >50K cells: subsample to 2-5K per cell type per condition. Do NOT use flat global cap | references/workflow-details.md |
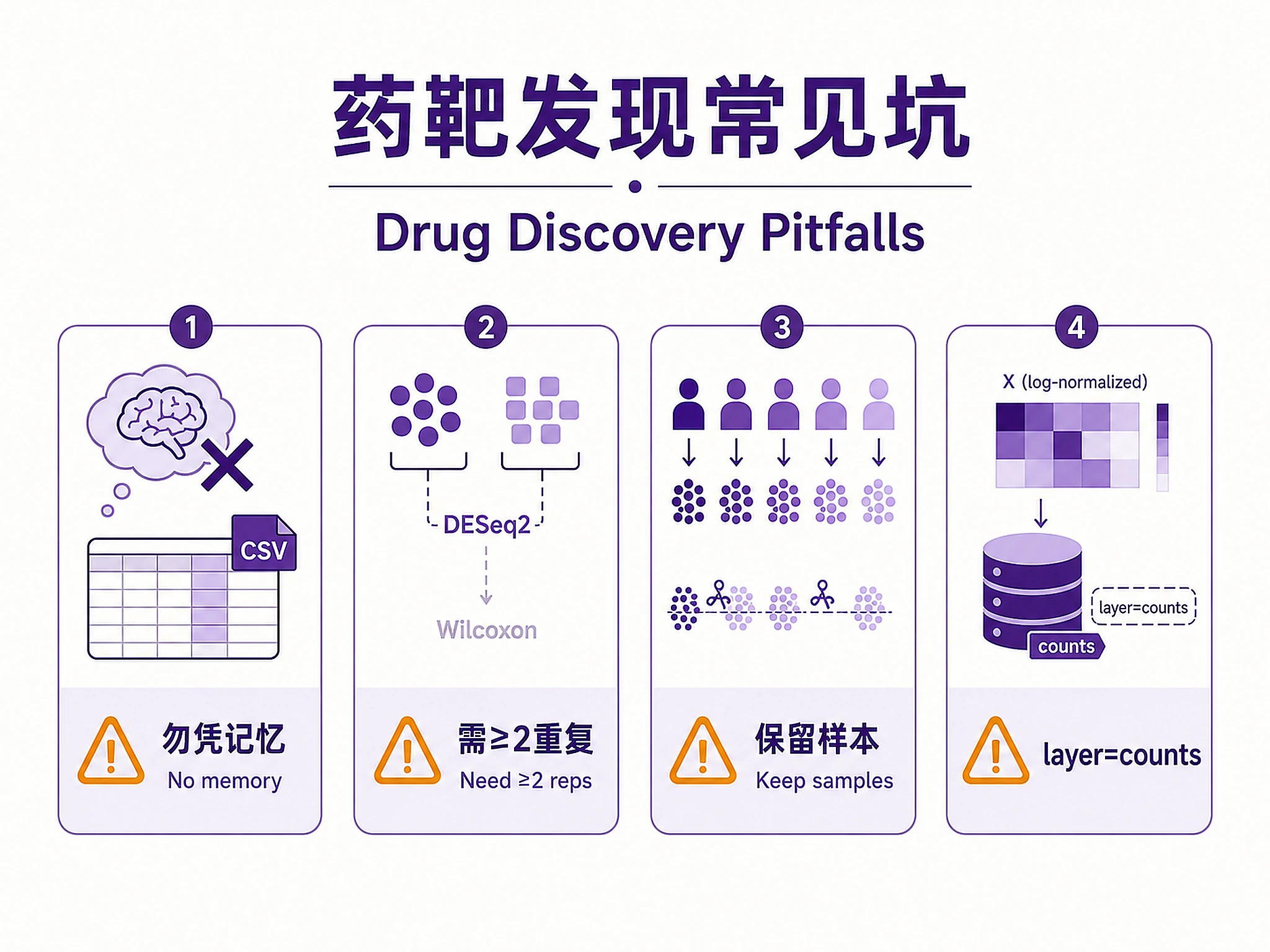
| DE layer | Always pass layer="counts" if .X is log-normalized. Auto-detected if layers["counts"] exists |
references/workflow-details.md |
| GSEA ranking | Combined stat sign(LFC) x -log10(padj) (default). Skips cell types with failed DE | references/workflow-details.md |
| Genetic evidence | Open Targets GWAS/ClinVar is primary; GeneBass if disease is in UKB; TWAS/eQTL supplementary | references/genetic_evidence_guide.md |
| Analysis scope | Full pipeline (default). Transcriptomics-only if no genetic evidence needed | Below |
Clarification Questions
Default settings (use unless user specifies otherwise):
- Species: Human | Genetic evidence: Enabled | All analyses: Enabled
1. Input Files (ASK THIS FIRST):
- Do you have an annotated scRNA-seq AnnData (.h5ad) with disease and control conditions?
- Or use demo data? GSE195452: Systemic sclerosis skin biopsies (SSc + healthy controls)
- If your own data: provide path to .h5ad file
2. Metadata columns (own data only):
- Which column contains cell type labels? (default:
cell_type) - Which column contains disease/condition labels? (default:
condition) - Which column contains sample/donor IDs? (default:
sample_id)
3. Disease context (own data only):
- What disease are you studying? (needed for Open Targets genetic evidence and pathway context)
- Species: Human (default) or Mouse?
4. Analysis scope:
- a) Full pipeline with genetic evidence (recommended)
- b) Transcriptomics only (DE + pathways + L-R, no genetic evidence)
- c) Custom: specify which analyses
:warning: IF DEMO DATA SELECTED: All parameters are pre-defined (SSc, human, full pipeline). DO NOT ask questions 2-4. Proceed directly to Step 1.
Data Integrity Rules
:rotating_light: NEVER reconstruct results from memory, notes, or session summaries. :rotating_light:
If you cannot find the expected output CSV files after a crash or restart:
- STOP immediately -- do NOT type quantitative values from memory
- Check standard locations:
results/,./results/,/mnt/results/, the working directory - Each analysis step saves CSVs incrementally (DE per cell type, GSEA per cell type, ranked_targets.csv) -- check for partial results
- If files are truly missing, re-run the analysis step from the saved h5ad
- NEVER hardcode target data like
[('GENE1', 0.82, 'HIGH'), ...]from session notes
Every quantitative value in the final report must be read from a CSV file on disk, not from session memory.
If the session crashed (OOM, kernel restart): Partial CSVs from completed steps are on disk. Re-run only the steps that didn't complete. The h5ad file is the checkpoint -- all analysis can be re-derived from it.
Standard Workflow
:rotating_light: MANDATORY: USE SCRIPTS EXACTLY AS SHOWN - DO NOT WRITE INLINE CODE :rotating_light:
Detailed step-by-step code: references/workflow-details.md
CRITICAL - DO NOT:
- Write inline analysis code -- STOP: Use the script functions
- Write custom export code -- STOP: Use
export_all() - Skip verification messages -- STOP: Check for verification messages after each step
- Reconstruct values from memory after a crash -- STOP: Read from saved CSVs or re-run
IF SCRIPTS FAIL - Script Failure Hierarchy:
- Fix and Retry (90%) - Install missing package, re-run script
- Modify Script (5%) - Edit the script file itself, document changes
- Use as Reference (4%) - Read script, adapt approach, cite source
- Write from Scratch (1%) - Only if genuinely impossible, explain why
NEVER skip directly to writing inline code without trying the script first.
Step 1 -- Load and validate data:
# Option A: Demo data (GSE195452 SSc)
from load_data import load_demo_ssc_data
adata = load_demo_ssc_data()
# Option B: Pre-annotated h5ad
from load_data import load_annotated_h5ad
adata = load_annotated_h5ad("path/to/adata.h5ad",
celltype_key="cell_type",
condition_key="condition",
sample_key="sample_id")
# Option C: Raw data (will preprocess)
from load_data import load_raw_data
from preprocess import run_preprocessing_pipeline
adata = load_raw_data("path/to/data")
adata = run_preprocessing_pipeline(adata, species="human")
# CHECKPOINT: Save AnnData to disk immediately (OOM/timeout resilience)
# If the kernel restarts, reload from this file instead of rebuilding.
adata.write_h5ad("results/adata_processed.h5ad")
print("Checkpoint saved: results/adata_processed.h5ad")
DO NOT write inline data loading code. CHECKPOINT: After loading, ALWAYS save the AnnData to disk. If the kernel restarts later, reload with sc.read_h5ad("results/adata_processed.h5ad") instead of re-downloading and rebuilding from GEO.
✅ VERIFICATION: You MUST see: "Data loaded successfully! N cells, M genes, K cell types, C conditions"
If you don't see this: Check file path, verify .h5ad has required metadata columns (cell_type, condition, sample_id).
Step 2 -- Run analysis:
Sample selection: Use ALL available samples — statistical power depends on biological replicates, not cell count. Subsample CELLS within samples if needed for memory, but do NOT drop samples. For datasets with >100K cells, subsample to ~3000-5000 cells per cell type per condition while keeping all samples represented. For plate-based data (Smart-seq2), use patient_id (not amplification batch) as the pseudobulk grouping variable. See references/workflow-details.md for details.
GSEA thresholds: GSEA output tables report all pathways with FDR < 0.25 (standard for GSEA reporting and exploration). Target scoring uses stricter FDR < 0.05 for pathway_score contribution. Both thresholds are intentional.
# 2a. Differential expression (disease vs control per cell type)
# IMPORTANT: pass layer="counts" if adata.X is log-normalized
from pseudobulk_de import run_pseudobulk_de, get_disease_signature_genes
de_results = run_pseudobulk_de(adata,
condition_key="condition",
sample_key="sample_id",
celltype_key="cell_type",
reference="Healthy",
layer="counts")
# 2b. Pathway enrichment (uses combined ranking: sign(LFC) x -log10(padj))
from pathway_enrichment import run_pathway_analysis
pathway_results = run_pathway_analysis(adata, de_results, species="human")
# 2c. Ligand-receptor interactions
# Auto-merges to max 20 cell types, subsamples to 2000 cells/type, 100 permutations
# for tractable runtime (~5-10 min). Increase n_perms for publication-quality.
from ligand_receptor import run_lr_analysis
lr_results = run_lr_analysis(adata,
celltype_key="cell_type",
condition_key="condition",
max_celltypes=20,
max_cells_per_type=2000,
n_perms=100)
# 2d. Collect candidate gene list from DE results (for genetic + OT queries)
sig_genes = get_disease_signature_genes(de_results, padj_threshold=0.05, log2fc_threshold=0.25)
candidate_genes = list(set(g for genes in sig_genes.values() for g in genes))[:200]
# 2e. Genetic evidence (queries Open Targets GWAS/ClinVar + TWAS + eQTL + L2G)
from genetic_evidence import collect_genetic_evidence
genetic_results = collect_genetic_evidence(
gene_list=candidate_genes,
disease_name="systemic sclerosis",
include_genebass=True)
# 2f. Target scoring
# CRITICAL: Use score_targets_simple() — DO NOT write custom scoring code.
# Custom scorers miss: convergence bonus, cross-compartment L-R bridging,
# adaptive reweighting, NES-weighted pathway scoring, and DE cap logic.
from target_scoring import score_targets_simple
scores = score_targets_simple({
"de": de_results,
"pathway": pathway_results,
"lr": lr_results,
"genetic": genetic_results,
})
# Druggability auto-queried for top 20 genes. For all: pass "ot_annotations" key.
# 2g. GWAS Gene Landscape (MANDATORY for SSc demo)
# Maps top SSc GWAS genes to their cell-type expression context.
# Separate from the DE-derived target list — bridges immune-fibroblast disconnect.
from genetic_evidence import get_top_gwas_genes, assess_gwas_genes_in_adata
gwas_genes = get_top_gwas_genes("systemic sclerosis", n=20)
gwas_context = assess_gwas_genes_in_adata(adata, gwas_genes, de_results=de_results)
DO NOT write custom scoring code. The score_targets_simple() function handles adaptive reweighting, convergence bonuses, cross-compartment L-R bridging, and NES-weighted pathway scoring. Custom scorers miss these and produce invalid priority tiers.
✅ VERIFICATION: After 2f you MUST see: "Target scoring complete! N targets scored, M HIGH, ..."
After 2g: "GWAS gene landscape: N genes mapped, M are DE in at least one cell type"
If DE returns 0 DEGs for a cell type: DESeq2 auto-falls back to Wilcoxon. GSEA quality gate skips cell types with failed DE.
If LIANA takes >15 min: Normal for large datasets. Partial results saved at each step.
Report structure: The PDF report should have TWO target sections:
- Multi-omics targets — DE-derived, scored across all evidence dimensions (from
score_targets) - GWAS gene landscape — top SSc genetic targets mapped to cell types (from
assess_gwas_genes_in_adata), showing which GWAS genes are expressed where and whether they're DE. This bridges the immune-fibroblast disconnect: GWAS targets (IRF5, STAT4) are immune genes, while DE targets are fibroblast genes. Both are real biology.
Step 3 -- Generate visualizations:
from generate_visualizations import generate_all_plots
generate_all_plots(adata, de_results, pathway_results, lr_results,
genetic_results, scores, output_dir="results",
gwas_context_df=gwas_context)
DO NOT write inline plotting code. This generates 11 standard visualizations including the GWAS gene landscape.
✅ VERIFICATION: You MUST see: "All plots generated successfully! N visualizations saved"
If a plot fails: Non-fatal -- other plots still generated. Check WARNING messages.
Step 4 -- Export results:
from export_results import export_all
export_all(adata, de_results, pathway_results, lr_results,
genetic_results, scores, output_dir="results")
DO NOT write custom export code. Use export_all().
✅ VERIFICATION: You MUST see: "=== Export Complete ==="
If you don't see "Export Complete": The export did not complete. Re-run the export function.
Target Scoring Methodology
See references/target_scoring_methodology.md for full details.
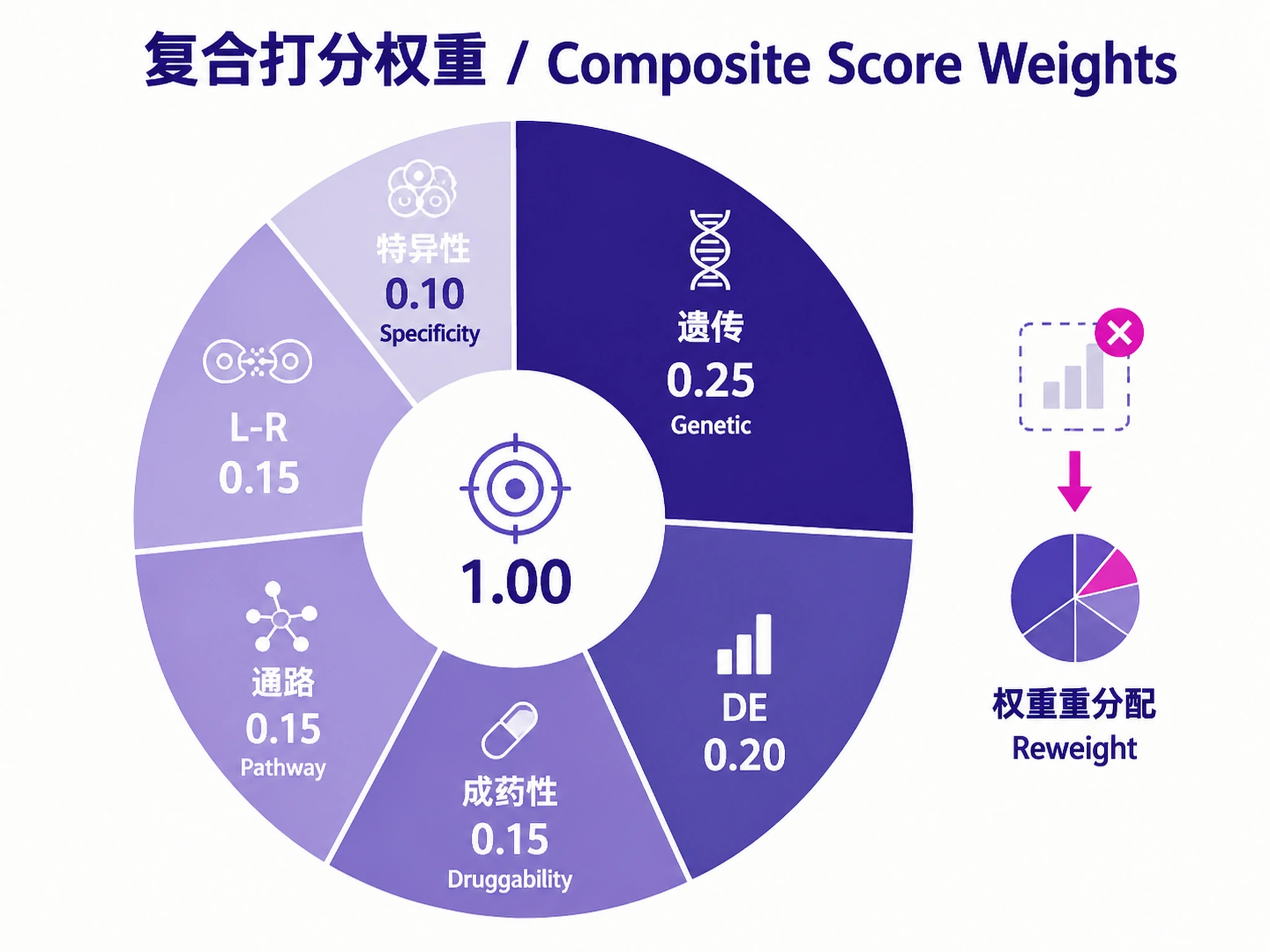
Multi-omics composite score (convergent genetic + transcriptomic evidence):
| Component | Weight | Description |
|---|---|---|
| Differential expression | 0.20 | -log10(padj) weighted by log2FC magnitude. Multi-cell-type DE bonus. |
| Pathway centrality | 0.15 | Disease-relevant enriched pathways containing the gene (FDR < 0.05). |
| L-R involvement | 0.15 | Consensus rank for disease-specific interactions. Reweighted to 0 if unavailable. |
| Cell-type specificity | 0.10 | Tau-like specificity of disease effect in disease-relevant cell types. |
| Genetic evidence | 0.25 | Open Targets GWAS/ClinVar (primary) + GeneBass + TWAS + eQTL + L2G. |
| Druggability | 0.15 | Open Targets tractability, known drugs, genetic constraint. |
Convergence bonus (1.2x): Targets with BOTH genetic AND transcriptomic evidence.
Priority tiers: HIGH (>=0.55), MEDIUM (0.35-0.55), LOW (<0.35)
Adaptive reweighting: If L-R or genetic evidence is unavailable, those weights are redistributed to remaining components (weights always sum to 1.0).
Common Issues
| Error | Cause | Solution |
|---|---|---|
ImportError: liana |
Not installed | pip install liana |
ImportError: decoupler |
Not installed | pip install decoupler |
| Pseudobulk DE blocked: N=1 | Single sample per condition | Need >=2 replicates. Fallback: cell-level Wilcoxon (exploratory) |
| DESeq2 degenerate results (all padj = 1.0) | Insufficient pseudobulk replicates for a cell type | Auto-falls back to Wilcoxon for that cell type. GSEA quality gate skips it. |
| GSEA skipped for a cell type | 0 significant DEGs (DE failure) | Expected -- GSEA on failed DE produces artifacts. Only runs on cell types with real signal. |
| GeneBass data not found | Not on Biomni datalake | Skill uses Open Targets GWAS/ClinVar as primary genetic evidence instead |
| No significant L-R interactions | Too few cell types or cells | Need >=3 cell types, >=10 cells per type. L-R weight redistributed in scoring. |
| LIANA takes >15 minutes | Large dataset with many cell types | Normal for >10K cells x 10+ cell types. Partial results saved at each step. |
| TWAS Atlas timeout | API issues | Retry with backoff (built-in). Falls back to other genetic evidence. |
| Open Targets rate limit | Too many gene queries | Built-in rate limiting (0.5s between requests) |
| SVG export failed | Missing svglite/cairocffi | Normal -- PNG always generated. Both created in most environments. |
Expected warnings (not errors):
| Warning | Meaning | Action |
|---|---|---|
| DESeq2 degenerate results, falling back to Wilcoxon | Cell type has too few samples for pseudobulk | Normal for subsampled data. Wilcoxon results are exploratory. |
| Skipping GSEA for [cell type] | DE failed for that cell type | GSEA quality gate working correctly. No action needed. |
| L-R consensus_rank is all NaN | liana version mismatch | Auto-falls back to specificity_rank. Scoring reweights. |
Suggested Next Steps
- Deep-dive on top targets -- Use
genetic-target-hypothesisfor detailed genetic characterization of top-scored targets - Trajectory analysis -- Use
scrna-trajectory-inferenceto trace disease progression in key cell types - Regulatory networks -- Use
grn-pyscenicto identify TF regulators of disease programs - Validation -- Use
literature-reviewto check published evidence for top targets
Related Skills
Upstream: scrnaseq-scanpy-core-analysis (produces input h5ad) | Complementary: genetic-target-hypothesis (genetics-first approach), cell-cell-communication (R/CellChat alternative), functional-enrichment-from-degs (R/clusterProfiler alternative) | Downstream: grn-pyscenic, scrna-trajectory-inference, literature-review
References
- Scanpy: Wolf FA, et al. (2018) Genome Biol. 19:15.
- PyDESeq2: Muzellec B, et al. (2023) Bioinformatics. 39(9):btad547.
- LIANA: Dimitrov D, et al. (2022) Nat Commun. 13:3224.
- decoupler: Badia-i-Mompel P, et al. (2022) Bioinformatics. 38(Supplement_2):ii81-ii88.
- GeneBass: Karczewski KJ, et al. (2022) Nature. 590:791-797.
- Open Targets: Ochoa D, et al. (2023) Nucleic Acids Res. 51(D1):D1302-D1310.
- TWAS Atlas: TWAS Atlas 2.0 (2025) Nucleic Acids Res.
Detailed guides: references/workflow-details.md | references/common-patterns.md | references/ssc_biology.md | references/target_scoring_methodology.md | references/genetic_evidence_guide.md | references/dataset_guide.md | references/liana_methods.md
Scripts: scripts/ | Evaluation: assets/eval/
Code preview
scripts/export_results.py
#!/usr/bin/env python3
"""
Export all analysis results for disease drug discovery pipeline.
Exports: annotated h5ad, DE CSVs, pathway CSVs, L-R CSVs, genetic evidence
CSVs, ranked target list, target evidence cards JSON.
Usage:
from export_results import export_all
export_all(adata, de_results, pathway_results, lr_results,
genetic_results, scores, output_dir="results")
"""
import json
import os
import sys
import numpy as np
import pandas as pd
def export_all(adata, de_results, pathway_results, lr_results,
genetic_results, scores_df, ot_annotations=None,
target_cards=None, output_dir="results"):
"""Export all analysis results to files.
Args:
adata: Annotated AnnData object
de_results: Dict of {celltype: DE DataFrame}
pathway_results: Dict with gsea_results, pathway_activity, disease_pathways
lr_results: Dict with interactions, disease_specific, network_summary
genetic_results: Dict from collect_genetic_evidence()
scores_df: DataFrame from score_targets()
ot_annotations: Dict from query_target_annotations()
target_cards: List from generate_target_cards()
output_dir: Output directory
Returns:
Dict of exported file paths
"""
os.makedirs(output_dir, exist_ok=True)
exported = {}
n_files = 0
print(f"Exporting results to {output_dir}/...")
# 1. Annotated AnnData
try:
h5ad_path = os.path.join(output_dir, "adata_analyzed.h5ad")
# Store analysis summary in .uns
adata.uns["drug_discovery_analysis"] = {
"n_celltypes_tested": len(de_results),
"n_targets_scored": len(scores_df) if scores_df is not None else 0,
"analysis_type": "disease_drug_discovery",
}
adata.write_h5ad(h5ad_path)
exported["h5ad"] = h5ad_path
n_files += 1
print(f" [{n_files}] AnnData: {h5ad_path}")
except Exception as e:
print(f" WARNING: H5AD export failed: {e}", file=sys.stderr)
# 2. Differential expression results
for celltype, df in de_results.items():
try:
safe_name = celltype.replace(" ", "_").replace("/", "-")
csv_path = os.path.join(output_dir, f"de_results_{safe_name}.csv")
df.to_csv(csv_path)
exported[f"de_{safe_name}"] = csv_path
n_files += 1
except Exception as e:
print(f" WARNING: DE export for {celltype} failed: {e}", file=sys.stderr)
# DE summary across cell types
try:
summary_rows = []
for ct, df in de_results.items():
padj_col = "padj" if "padj" in df.columns else "pvals_adj"
fc_col = "log2FoldChange" if "log2FoldChange" in df.columns else "logfoldchanges"
if padj_col in df.columns:scripts/generate_visualizations.py
#!/usr/bin/env python3
"""
Generate publication-quality visualizations for disease drug discovery.
All plots are saved in both PNG (300 DPI) and SVG formats.
Usage:
from generate_visualizations import generate_all_plots
generate_all_plots(adata, de_results, pathway_results, lr_results,
genetic_results, scores, output_dir="results")
"""
import os
import sys
import warnings
import numpy as np
import pandas as pd
warnings.filterwarnings("ignore", category=FutureWarning)
def _setup_matplotlib():
"""Configure matplotlib for publication-quality output."""
import matplotlib
matplotlib.use("Agg")
import matplotlib.pyplot as plt
import seaborn as sns
sns.set_style("ticks")
plt.rcParams.update({
"figure.dpi": 300,
"savefig.dpi": 300,
"font.size": 10,
"axes.titlesize": 12,
"axes.labelsize": 10,
"figure.figsize": (8, 6),
})
return plt, sns
def _save_plot(fig, output_dir, name):
"""Save a figure in both PNG and SVG formats."""
os.makedirs(output_dir, exist_ok=True)
png_path = os.path.join(output_dir, f"{name}.png")
fig.savefig(png_path, dpi=300, bbox_inches="tight", facecolor="white")
try:
svg_path = os.path.join(output_dir, f"{name}.svg")
fig.savefig(svg_path, format="svg", bbox_inches="tight", facecolor="white")
except Exception:
pass # SVG export is optional
import matplotlib.pyplot as plt
plt.close(fig)
return png_path
# ---------------------------------------------------------------------------
# Individual plot functions
# ---------------------------------------------------------------------------
def plot_umap_overview(adata, output_dir="results"):
"""Plot UMAP colored by cell type and condition."""
plt, sns = _setup_matplotlib()
import scanpy as sc
fig, axes = plt.subplots(1, 2, figsize=(16, 7))
# Cell type UMAP
if "X_umap" in adata.obsm:
sc.pl.umap(adata, color="cell_type", ax=axes[0], show=False,
title="Cell Types", frameon=True)
sc.pl.umap(adata, color="condition", ax=axes[1], show=False,
title="Condition", frameon=True)
else:
axes[0].text(0.5, 0.5, "UMAP not computed", ha="center", va="center",
transform=axes[0].transAxes)
axes[1].text(0.5, 0.5, "UMAP not computed", ha="center", va="center",
transform=axes[1].transAxes)
scripts/genetic_evidence.py
#!/usr/bin/env python3
"""
Integrate genetic evidence from multiple sources for disease-gene prioritization.
Queries four complementary genetic evidence databases and integrates results
into a unified per-gene genetic evidence summary:
1. GeneBass — rare variant burden test results (pLoF, missense)
2. TWAS Atlas (CNCB-NGDC) — published TWAS associations across traits/tissues
3. EBI eQTL Catalogue — tissue-specific eQTL associations
4. Open Targets L2G — locus-to-gene GWAS prioritization scores
The combined evidence feeds into a genetic sub-score used for target
prioritization in the scRNA-seq disease-drug discovery workflow.
Usage:
python genetic_evidence.py --genes "IRF4,STAT4,BLK,TNFAIP3" --disease "systemic sclerosis"
python genetic_evidence.py --genes "IRF4,STAT4" --disease "systemic sclerosis" --output genetic_evidence.json
python genetic_evidence.py --gene-file gene_list.txt --disease "scleroderma" --no-genebass
"""
import argparse
import json
import os
import sys
import time
import numpy as np
import pandas as pd
try:
import requests
except ImportError:
print("ERROR: requests library required. Install with: pip install requests",
file=sys.stderr)
sys.exit(1)
import urllib.parse
import urllib.request
# ---------------------------------------------------------------------------
# Constants
# ---------------------------------------------------------------------------
TWAS_ATLAS_BASE = "https://ngdc.cncb.ac.cn/twas"
EQTL_API_BASE = "https://www.ebi.ac.uk/eqtl/api/v2"
OT_GRAPHQL = "https://api.platform.opentargets.org/api/v4/graphql"
REQUEST_DELAY = 0.5
MAX_RETRIES = 3
# Tissues relevant to SSc / autoimmune / fibrotic disease
EQTL_SKIN_IMMUNE_TISSUES = {
"skin", "skin - sun exposed (lower leg)", "skin - not sun exposed (suprapubic)",
"whole blood", "blood", "lymphocytes", "monocytes", "neutrophils",
"T cells", "CD4+ T cells", "CD8+ T cells", "B cells", "NK cells",
"macrophages", "dendritic cells", "LCL", "lymphoblastoid cell line",
"PBMC", "spleen", "lung", "fibroblast", "fibroblasts",
}
# TWAS trait search terms — disease-specific only, no proxy diseases
SSC_RELATED_TRAITS = [
"systemic sclerosis",
"scleroderma",
"pulmonary fibrosis",
"interstitial lung disease",
"Raynaud",
]
# ---------------------------------------------------------------------------
# Shared helpers
# ---------------------------------------------------------------------------
def _retry_request(func, *args, retries=MAX_RETRIES, delay=REQUEST_DELAY, **kwargs):
"""Execute a callable with exponential-backoff retry logic."""
for attempt in range(retries):
try:
result = func(*args, **kwargs)Companion files
| Type | Path | Bytes |
|---|---|---|
| Markdown | references/common-patterns.md | 2,996 |
| Markdown | references/dataset_guide.md | 2,092 |
| Markdown | references/genetic_evidence_guide.md | 3,781 |
| Markdown | references/liana_methods.md | 1,730 |
| Markdown | references/ssc_biology.md | 4,753 |
| Markdown | references/target_scoring_methodology.md | 4,112 |
| Markdown | references/workflow-details.md | 6,742 |
| Python | scripts/export_results.py | 10,851 |
| Python | scripts/generate_visualizations.py | 24,038 |
| Python | scripts/genetic_evidence.py | 50,299 |
| Python | scripts/ligand_receptor.py | 33,315 |
| Python | scripts/load_data.py | 16,982 |
| Python | scripts/pathway_enrichment.py | 26,058 |
| Python | scripts/preprocess.py | 16,717 |
| Python | scripts/pseudobulk_de.py | 25,263 |
| Python | scripts/query_opentargets.py | 8,600 |
| Python | scripts/target_scoring.py | 33,823 |
| Markdown | SKILL.md | 23,046 |
| JSON | skill.meta.json | 3,860 |