GWAS to Function (TWAS)
From GWAS to causal genes via TWAS, colocalization and MR.
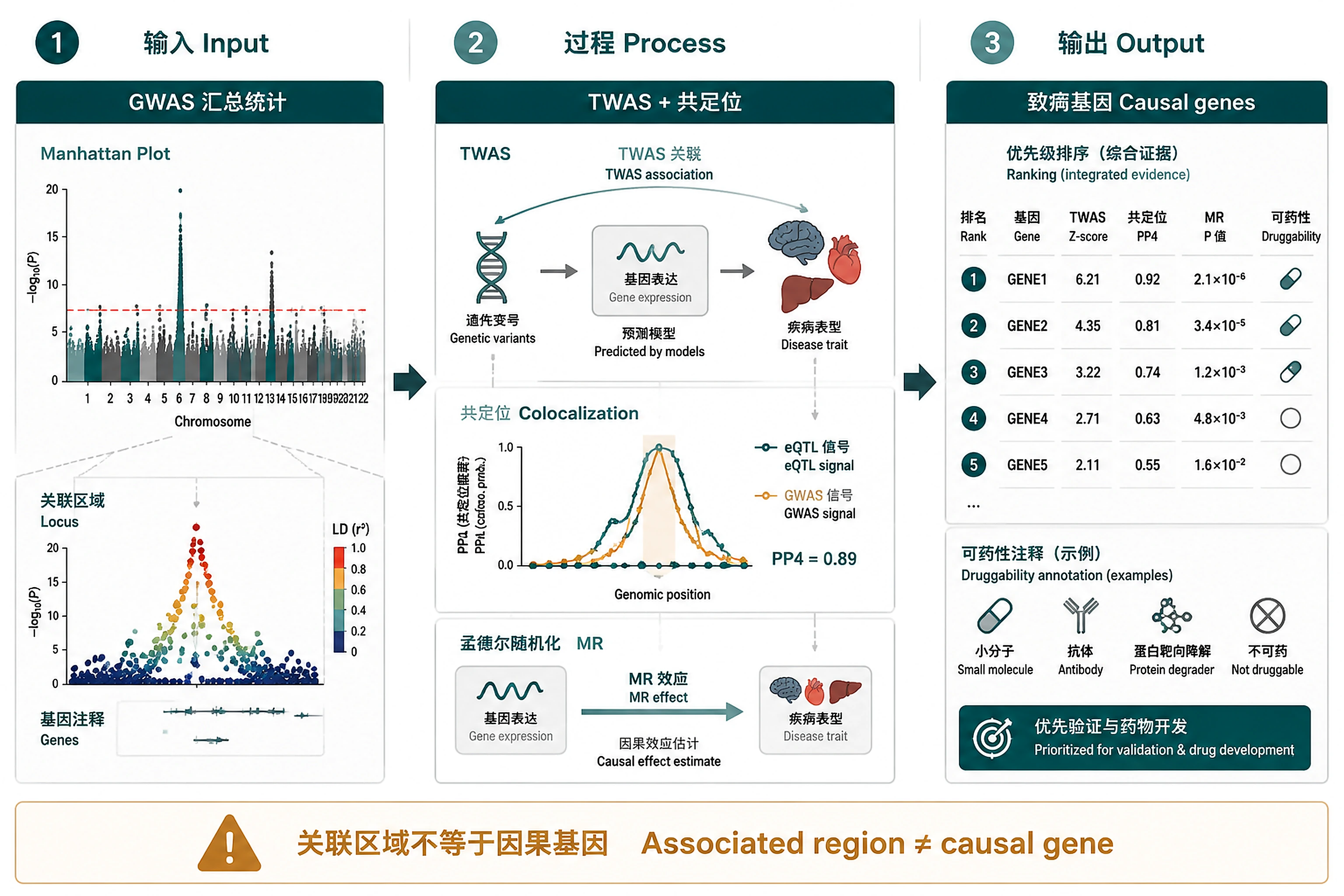
Overview
Problem. GWAS flags regions; LD hides the actual causal gene.
Learning goals
- From associated region to causal gene
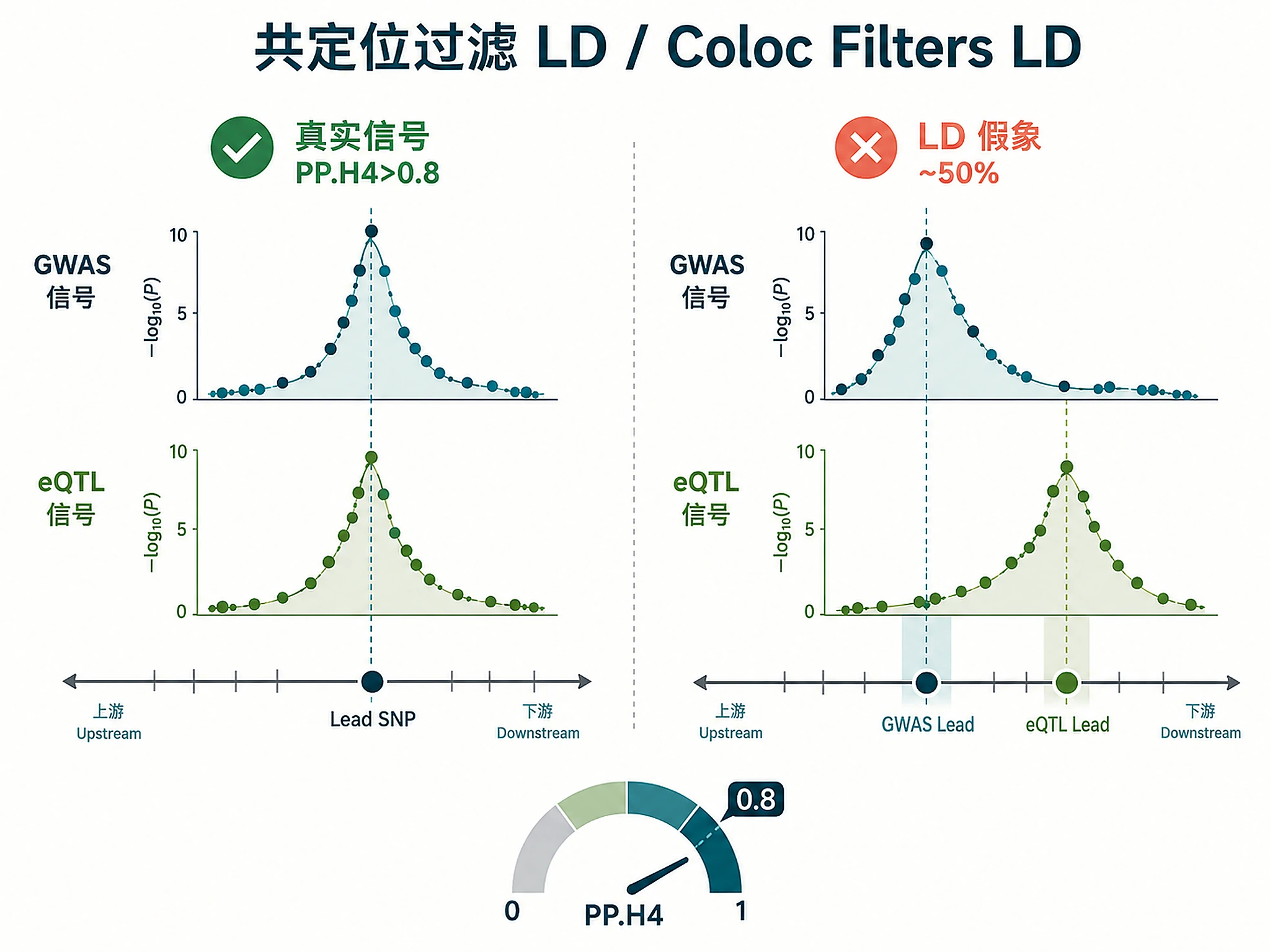
- Coloc separates cause from LD artefact
Figures
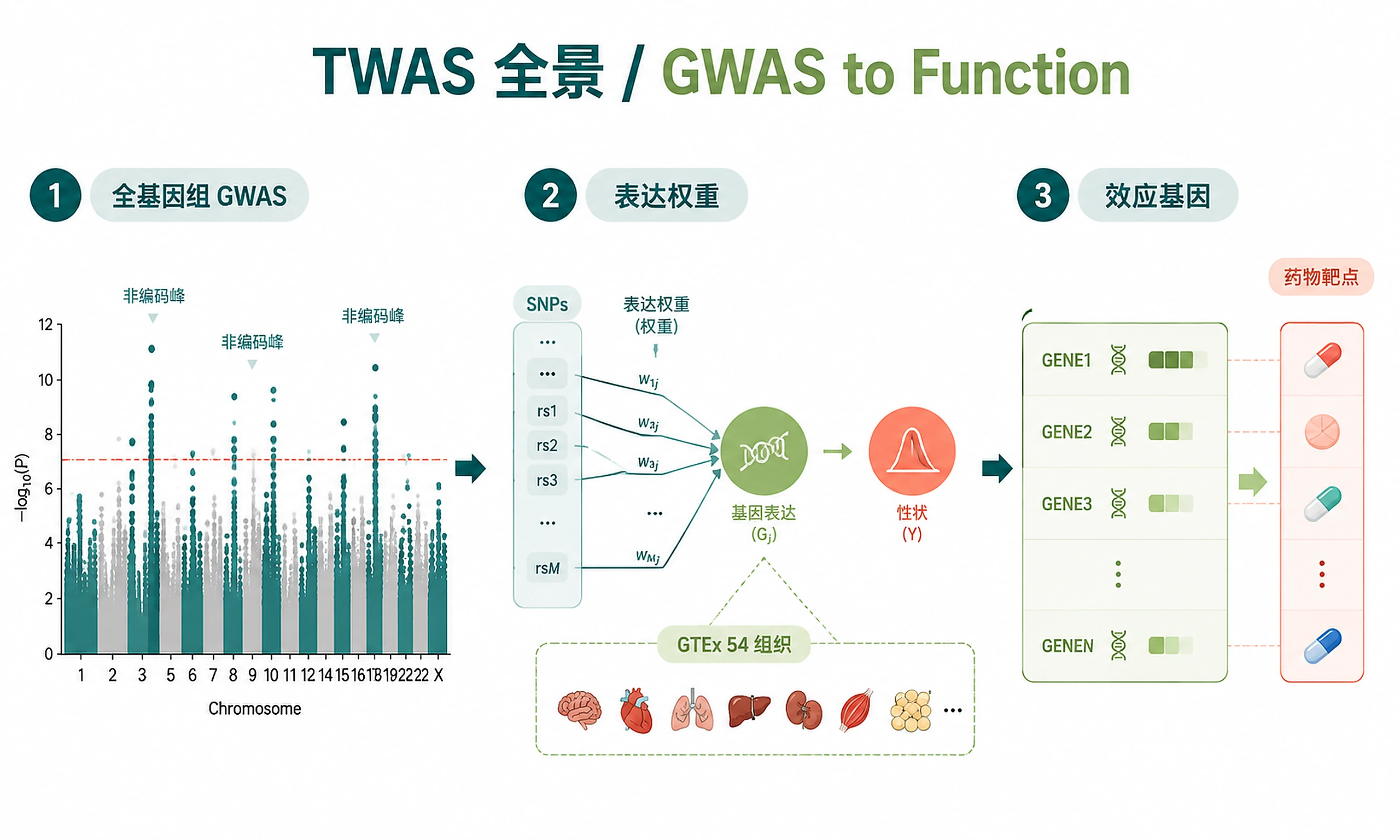
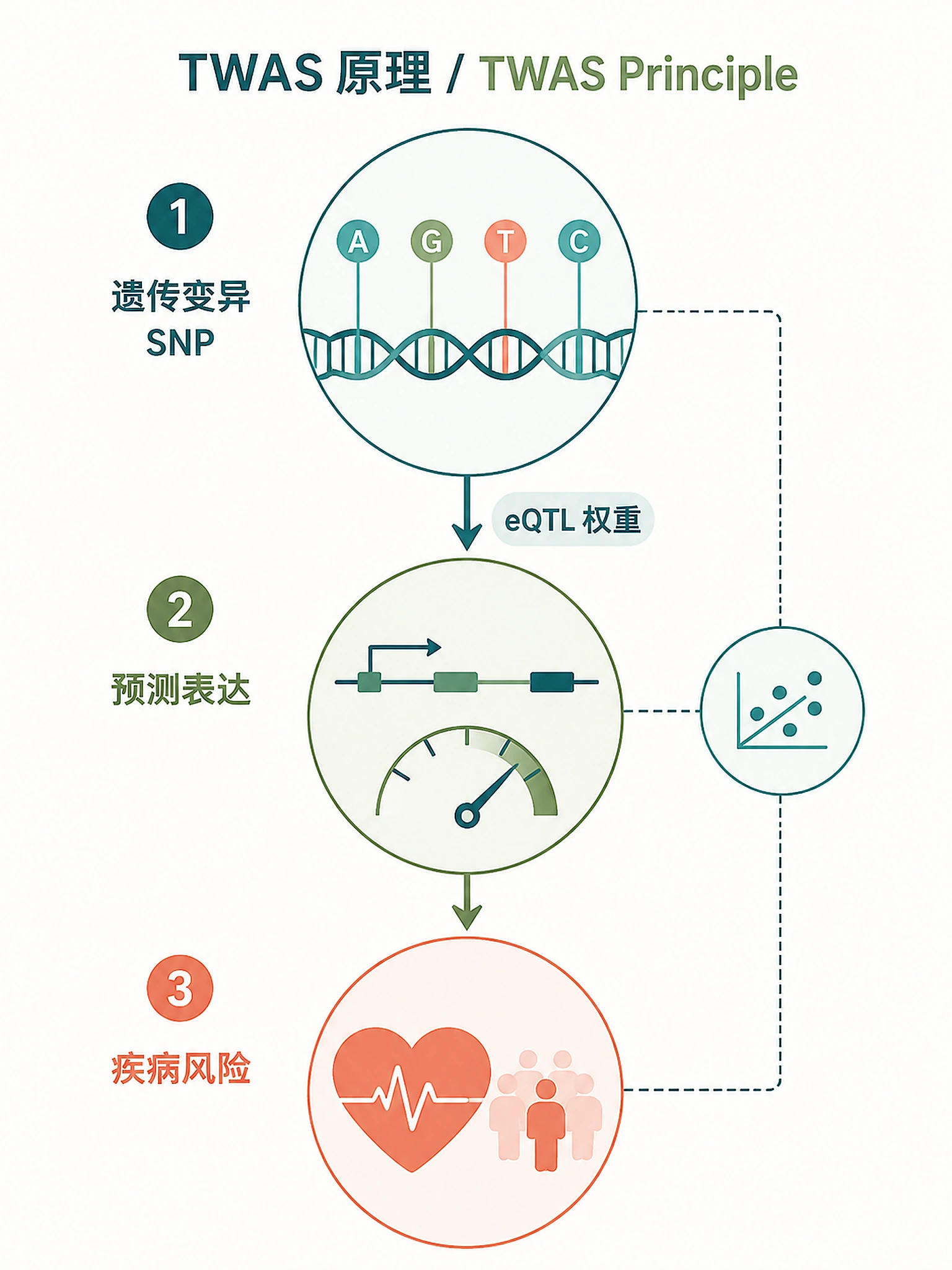
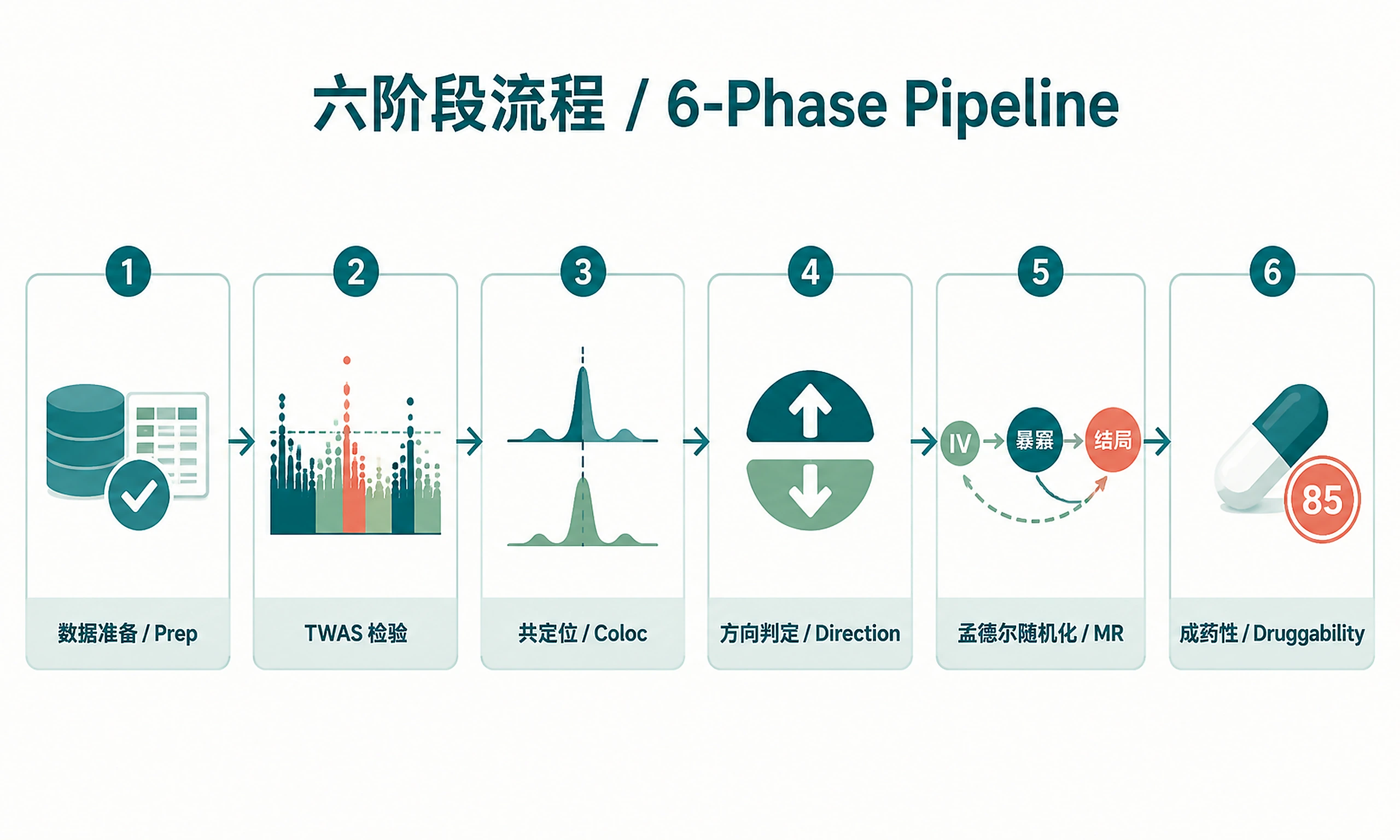

Tutorial
Identify genes whose genetically regulated expression is associated with disease risk, determine therapeutic directionality (inhibit vs. activate), and prioritize drug targets with causal genetic evidence using Transcriptome-Wide Association Study (TWAS) analysis.
Key capabilities:
- Dual TWAS tools: FUSION (comprehensive) and S-PrediXcan (fast, 10-100x faster)
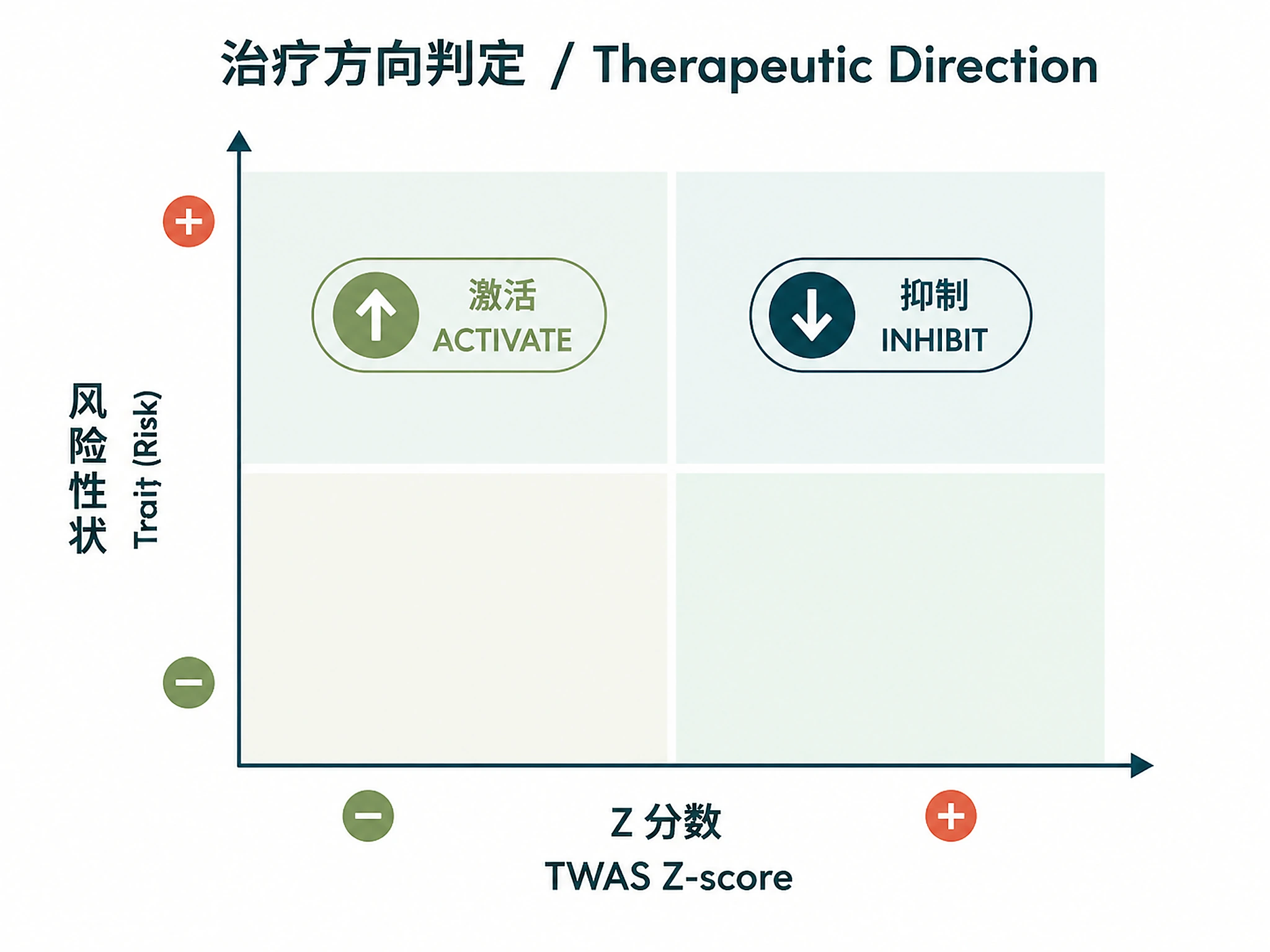
- Therapeutic directionality: Determine inhibit vs. activate strategy for each gene
- Multi-tier analysis: Basic → Colocalization → Mendelian Randomization → Druggability
- Colocalization testing: Filters LD artifacts (up to 50% of TWAS hits), requires PP.H4 > 0.8
- Cross-tissue meta-analysis: S-MultiXcan across 54 GTEx v8 tissues
- Biopharma-ready reports: Excel workbooks with prioritized targets and confidence levels
When to Use This Skill
Use this skill when:
- ✅ You have genome-wide GWAS summary statistics (N > 5,000 samples)
- ✅ Need to identify effector genes for non-coding GWAS variants
- ✅ Want to determine therapeutic strategy (inhibit vs. activate gene expression)
- ✅ Prioritizing drug targets by genetic evidence strength
- ✅ Validating existing targets with causal genetic evidence
Don't use this skill when:
- ❌ Only have significant loci (p < 5×10⁻⁸) without genome-wide data
- ❌ Sample size < 5,000 (insufficient power for TWAS)
- ❌ Need functional enrichment analysis → use functional-enrichment-from-degs
- ❌ Working with individual-level genotypes → run GWAS first with gwas-genotype-qc
Data requirements: Genome-wide GWAS summary statistics with columns: SNP, CHR, BP, A1, A2, BETA/OR, SE, P, N
Installation
Required Software
| Software | Version | License | Commercial Use | Installation |
|---|---|---|---|---|
| FUSION | Latest | MIT | ✅ Permitted | https://github.com/gusevlab/fusion_twas |
| MetaXcan/S-PrediXcan | Latest | MIT | ✅ Permitted | pip install metaxcan |
| Python | ≥3.9 | PSF | ✅ Permitted | conda/pip |
| R | ≥4.0 | GPL | ✅ Permitted | CRAN (required for FUSION only) |
Python Packages
pip install pandas numpy scipy plotnine plotnine-prism seaborn statsmodels metaxcan adjustText
For detailed installation: See references/installation-guide.md for FUSION setup, LD reference panels, and GTEx weight downloads.
License compliance: All tools permit commercial AI agent use.
Inputs
Required:
- GWAS summary statistics: Genome-wide (not just significant loci)
- Format: Tab or comma-delimited text (gzip supported)
- Required columns: SNP, CHR, BP, A1, A2, BETA/OR, SE, P, N
- Minimum sample size: N > 5,000 (recommended N > 10,000)
- Genome build: GRCh37/hg19 or GRCh38 (must specify)
Optional:
- Trait information: Type (disease/quantitative), directionality (higher = risk/protective)
- Tissue selection: LDSC heritability enrichment results (from gwas-heritability-ldsc)
- eQTL summary statistics: For colocalization (GTEx v8 available via download)
Outputs
Tier 1 (Basic): TWAS results per tissue, Manhattan/QQ plots (PNG/SVG)
Tier 2 (+ Coloc): PP.H4 scores for each gene, therapeutic recommendations (inhibit/activate)
Tier 3 (+ MR): Causal estimates with sensitivity analyses, MR summary Excel
Tier 4 (+ Drug): reports/[trait]_Target_Report.xlsx - Comprehensive Excel workbook with executive summary, TWAS results, colocalization evidence, therapeutic directionality, MR causal inference, and druggability scores
Clarification Questions
Ask these questions before starting analysis:
-
Input Files (ASK THIS FIRST):
- Do you have specific GWAS summary statistics file(s) to analyze?
- If uploaded: Can you confirm this is genome-wide summary statistics (not just significant loci)?
- Expected format: SNP, CHR, BP, A1, A2, BETA/OR, SE, P, N columns
- Or use example/demo data? Example GWAS datasets can be downloaded for testing purposes
- Minimum requirements: N > 5,000 samples, genome-wide coverage
-
Trait Information:
- Trait type: Disease (binary) or quantitative trait?
- Trait directionality: Is higher value "risk" or "protective"?
- Sample size and ancestry composition?
- Trait category: Cardiovascular, metabolic, neurological, immune, cancer, etc.?
-
Tissue Selection Strategy:
- Data-driven (LDSC): Run LDSC partitioned heritability first (most rigorous, see gwas-heritability-ldsc)
- Biology-based: Use known trait-tissue relationships (see references/tissue_reference_guide.md)
- Comprehensive: Test all 54 GTEx tissues via S-MultiXcan (discovery mode)
-
Analysis Tier (choose depth based on goals and confidence requirements):
- Tier 1 - Basic TWAS: Association testing only, low-medium confidence
- Tier 2 - + Colocalization (RECOMMENDED): Filters LD artifacts (PP.H4 > 0.8), medium-high confidence
- Tier 3 - + Mendelian Randomization: Establishes causality, high confidence
- Tier 4 - + Druggability: Full target prioritization report, very high confidence (gold standard)
-
TWAS Tool Selection:
- FUSION (comprehensive): Multiple prediction models, built-in colocalization, requires R + >32GB RAM
- S-PrediXcan (fast): MASHR model, 10-100x faster than FUSION, requires Python + <16GB RAM
- See references/fusion_best_practices.md and references/spredixxcan_best_practices.md
-
Computational Resources:
- Local machine (<16GB RAM): S-PrediXcan, analyze <5 tissues
- Server/HPC (>32GB RAM): FUSION or S-PrediXcan, parallelize across all tissues
Standard Workflow
🚨 EXECUTE EXACTLY AS SHOWN - Do not modify these commands.
CRITICAL: Use relative paths (scripts/, references/). DO NOT construct absolute paths like /mnt/knowhow/ or /workspace/.
IMPORTANT: Call scripts via command line. DO NOT import and write inline Python code.
Phase 1: Data Preparation
Step 1: Validate and harmonize GWAS summary statistics
python scripts/validate_gwas_sumstats.py \
--gwas gwas_sumstats.txt.gz \
--min-n 5000 \
--build GRCh37 \
--output gwas_harmonized.txt
QC checks: Duplicate SNPs, allele harmonization, genomic inflation (λGC), LDSC intercept. For LDSC QC interpretation, see references/ldsc_qc_guidelines.md.
Step 2: Select tissues for TWAS analysis
Option A - Data-driven (after running gwas-heritability-ldsc):
python scripts/select_reference_panel.py \
--ldsc-results ../gwas-heritability-ldsc/twas_tissue_recommendations.csv \
--output selected_tissues.txt
Option B - Biology-based:
python scripts/select_reference_panel.py \
--trait-category cardiovascular \
--top-n 5 \
--output selected_tissues.txt
See references/tissue_reference_guide.md for trait-tissue mappings.
Step 3: Download expression weights and LD reference panels
See references/installation-guide.md for:
- GTEx v8 weight download links (FUSION: http://gusevlab.org/projects/fusion/, S-PrediXcan: https://predictdb.org/)
- LD reference panel setup (1000 Genomes EUR or FUSION LDREF)
Phase 2: TWAS Association Testing
Step 4: Run TWAS analysis
Option A - FUSION (comprehensive):
python scripts/run_fusion.py \
--gwas gwas_harmonized.txt \
--weights weights/GTEx_v8/ \
--tissues selected_tissues.txt \
--ref-ld-panel LDREF/ \
--output results/fusion/
Option B - S-PrediXcan (fast):
python scripts/run_spredixxcan.py \
--gwas gwas_harmonized.txt \
--weights weights/GTEx_v8/ \
--tissues selected_tissues.txt \
--output results/spredixxcan/
See references/fusion_best_practices.md and references/spredixxcan_best_practices.md for tool-specific parameters.
Step 5: Generate visualization plots
python scripts/plot_twas_results.py \
--twas-results results/fusion/Artery_Coronary_twas.txt \
--gwas gwas_harmonized.txt \
--output-dir figures/
Creates Manhattan plots, QQ plots, and regional association plots (PNG + SVG formats).
Step 5b (Optional): Validate against published TWAS results
python scripts/validate_with_twas_hub.py \
--twas-results results/fusion/Artery_Coronary_twas.txt \
--trait coronary_artery_disease \
--tissue Artery_Coronary
See references/twas_hub_validation_guide.md.
Phase 3: Statistical Refinement (Tier 2+)
Step 6: Colocalization analysis to filter LD artifacts
python scripts/colocalization_analysis.py \
--twas-results results/fusion/significant_genes.txt \
--gwas gwas_harmonized.txt \
--eqtl-dir eqtl_data/GTEx_v8/ \
--tissue Artery_Coronary \
--pp4-threshold 0.8 \
--output coloc_results/
CRITICAL: Up to 50% of TWAS hits may be LD artifacts. Only genes with PP.H4 > 0.8 have strong colocalization evidence.
Step 6b (Optional): Cross-tissue meta-analysis
python scripts/run_smultixcan.py \
--gwas gwas_harmonized.txt \
--weights weights/GTEx_v8/ \
--output results/smultixcan/
Phase 4: Therapeutic Directionality
Step 7: Interpret therapeutic strategy (inhibit vs. activate)
python scripts/interpret_therapeutic_direction.py \
--coloc-results coloc_results/high_confidence.txt \
--trait-direction risk \
--trait-name "Coronary Artery Disease" \
--output therapeutic_recommendations.csv
Logic: Positive TWAS Z-score + risk trait → INHIBIT gene; Negative Z-score + risk trait → ACTIVATE gene.
See references/therapeutic_interpretation_guide.md for comprehensive directionality framework.
Step 8: Multi-layer consistency check (eQTL + TWAS directionality)
python scripts/multilayer_direction_analysis.py \
--therapeutic-recs therapeutic_recommendations.csv \
--gwas gwas_harmonized.txt \
--eqtl-data eqtl_data/GTEx_v8/ \
--twas-results results/fusion/ \
--coloc-results coloc_results/ \
--output multilayer_consistency.csv
Confidence levels: High (consistent + PP.H4 > 0.8), Medium (PP.H4 = 0.5-0.8), Low (inconsistent/poor coloc).
Phase 5: Causal Validation (Tier 3+)
Step 9: Mendelian Randomization for causal inference
python scripts/mendelian_randomization.py \
--genes therapeutic_recommendations.csv \
--exposure-data eqtl_data/GTEx_v8/ \
--outcome-data gwas_harmonized.txt \
--methods ivw,egger,weighted_median \
--output mr_results/
MR establishes causal directionality (gene expression → trait). Includes sensitivity analyses for pleiotropy.
See references/mendelian_randomization_guide.md for MR principles and interpretation.
Phase 6: Druggability Assessment (Tier 4)
Step 10: Druggability scoring
python scripts/druggability_scoring.py \
--genes multilayer_consistency.csv \
--include-known-drugs \
--output druggability_scores.csv
Scores based on: Protein class, known drugs, clinical precedent, assay availability.
Step 11: Generate comprehensive target report
python scripts/export_results.py \
--twas-results results/fusion/ \
--coloc-results coloc_results/ \
--therapeutic-recs therapeutic_recommendations.csv \
--mr-results mr_results/ \
--druggability druggability_scores.csv \
--trait "Coronary Artery Disease" \
--output reports/CAD_Target_Report.xlsx
Creates publication-ready Excel workbook with executive summary, evidence layers, and prioritized targets.
That's it! The scripts handle everything automatically.
⚠️ CRITICAL - DO NOT:
- ❌ Write inline TWAS code → causes model mismatches, missing QC steps
- ❌ Write inline colocalization code → incorrect PP.H4 calculations, wrong priors
- ❌ Use absolute paths like
/mnt/knowhow/workflows/→ use relative pathsscripts/exactly as shown - ❌ Import script functions directly → use command-line interface for proper error handling and validation
- ❌ Skip phases based on tier → breaks dependency chain (e.g., Phase 4 requires Phase 3 colocalization results)
Phase dependencies:
- Phase 3 (Colocalization) requires Phase 2 TWAS results
- Phase 4 (Directionality) requires Phase 3 PP.H4 > 0.8 filtering
- Phase 5 (MR) requires Phase 4 directional interpretations
- Phase 6 (Druggability) requires Phase 5 causal evidence
DO NOT skip phases or run out of order.
Common Issues
| Issue | Cause | Solution |
|---|---|---|
| Genomic inflation (λGC > 1.1) | Population stratification or polygenicity | Run LDSC intercept test, check ratio < 0.15. See references/ldsc_qc_guidelines.md |
| No significant genes | Insufficient power, wrong tissues, or no true signals | Try FDR correction instead of Bonferroni, verify tissue relevance, check GWAS has genome-wide significant hits |
| Poor colocalization (PP.H4 < 0.5) | LD artifacts (common, affects ~50% of TWAS hits) | Expected behavior - filter these genes out, keep only PP.H4 > 0.8 |
| Allele mismatch errors | Strand flips, allele coding differences | Use harmonize_alleles() in Step 1, remove ambiguous A/T and C/G SNPs |
| FUSION memory errors | Insufficient RAM for large chromosomes | Use S-PrediXcan instead (10-100x less memory), or run chromosome-by-chromosome |
For complete troubleshooting guide, see references/troubleshooting_guide.md.
Suggested Next Steps
After completing TWAS analysis:
- Functional validation: Cross-reference with expression data, protein-protein interaction networks
- Pathway enrichment: Use functional-enrichment-from-degs on prioritized genes
- Variant annotation: Link causal variants to regulatory elements with genetic-variant-annotation
- Portfolio prioritization: Rank targets by genetic evidence strength, druggability, and clinical precedent
- Literature review: Check PubMed for supporting functional studies on top genes
Related Skills
Prerequisites:
- gwas-genotype-qc - Quality control for raw genotype data
- gwas-heritability-ldsc - Tissue enrichment analysis for tissue selection
Downstream analyses:
- functional-enrichment-from-degs - Pathway analysis for gene lists
- genetic-variant-annotation - Functional annotation of causal variants
Alternatives:
- SMR (Summary-data-based MR) - Alternative TWAS approach with built-in MR
- FOCUS (Fine-mapping Of CaUsal gene Sets) - Fine-mapping extension for TWAS
References
Internal documentation:
- references/ldsc_qc_guidelines.md - LDSC intercept interpretation
- references/tissue_reference_guide.md - Trait-tissue mappings (GTEx v8)
- references/fusion_best_practices.md - FUSION usage guidelines
- references/spredixxcan_best_practices.md - S-PrediXcan/S-MultiXcan guidelines
- references/therapeutic_interpretation_guide.md - Directionality → drug target framework
- references/mendelian_randomization_guide.md - MR principles and assumptions
- references/twas_hub_validation_guide.md - Comparison with published TWAS
- references/troubleshooting_guide.md - Common issues and solutions
- references/installation-guide.md - Detailed setup instructions
Key papers:
- FUSION: Gusev A, et al. (2016) Nat Genet 48:245-252. https://doi.org/10.1038/ng.3506
- S-PrediXcan: Barbeira AN, et al. (2018) Nat Commun 9:1825. https://doi.org/10.1038/s41467-018-03621-1
- S-MultiXcan: Barbeira AN, et al. (2019) PLoS Genet 15:e1007889
- Colocalization: Giambartolomei C, et al. (2014) PLoS Genet 10:e1004383
- MR: Hemani G, et al. (2018) eLife 7:e34408
Online resources:
- FUSION: http://gusevlab.org/projects/fusion/
- MetaXcan: https://github.com/hakyimlab/MetaXcan
- PredictDB: https://predictdb.org/ (GTEx v8 weights)
- GTEx Portal: https://gtexportal.org/
- TWAS Hub: http://twas-hub.org/ (published TWAS results)
- MR-Base: https://www.mrbase.org/
Code preview
scripts/colocalization_analysis.py
"""
Colocalization Analysis for TWAS
This module provides functions for colocalization testing to distinguish true
causal genes from LD artifacts using COLOC and related methods.
"""
import pandas as pd
import subprocess
from pathlib import Path
def run_coloc_for_twas_hits(twas_results, gwas_sumstats, eqtl_dir, tissue,
window_kb=500, output_dir="results/colocalization/"):
"""
Run colocalization analysis for significant TWAS genes.
Parameters
----------
twas_results : pandas.DataFrame
TWAS results with significant genes
gwas_sumstats : pandas.DataFrame or str
GWAS summary statistics
eqtl_dir : str
Directory containing eQTL summary statistics
tissue : str
Tissue name
window_kb : int
Window size around gene for colocalization (default: 500kb)
output_dir : str
Output directory
Returns
-------
pandas.DataFrame
Colocalization results with PP.H4 posterior probabilities
"""
output_path = Path(output_dir)
output_path.mkdir(parents=True, exist_ok=True)
print(f"Running colocalization analysis for {len(twas_results)} TWAS genes...")
coloc_results = []
for idx, row in twas_results.iterrows():
gene = row['GENE']
chrom = row['CHR']
gene_start = row['P0']
gene_end = row['P1']
# Define colocalization window
window_start = max(0, gene_start - window_kb * 1000)
window_end = gene_end + window_kb * 1000
print(f" {gene} (chr{chrom}:{window_start}-{window_end})")
# Extract GWAS data in window
if isinstance(gwas_sumstats, pd.DataFrame):
gwas_window = gwas_sumstats[
(gwas_sumstats['CHR'] == chrom) &
(gwas_sumstats['BP'] >= window_start) &
(gwas_sumstats['BP'] <= window_end)
]
else:
# Load from file
gwas_window = pd.read_csv(gwas_sumstats, sep='\t')
gwas_window = gwas_window[
(gwas_window['CHR'] == chrom) &
(gwas_window['BP'] >= window_start) &
(gwas_window['BP'] <= window_end)
]
# Extract eQTL data
eqtl_file = f"{eqtl_dir}/{tissue}/{gene}.eqtl.txt"
try:
eqtl_data = pd.read_csv(eqtl_file, sep='\t')
except FileNotFoundError:
print(f" WARNING: eQTL data not found for {gene}")
continue
scripts/druggability_scoring.py
"""
Druggability Assessment for TWAS Targets
This module provides functions for evaluating therapeutic feasibility of TWAS genes
based on protein class, known drugs, and structural features.
"""
import pandas as pd
# Druggable protein classes
TIER1_CLASSES = ['kinase', 'gpcr', 'ion channel', 'nuclear receptor', 'protease']
TIER2_CLASSES = ['enzyme', 'transporter', 'peptidase', 'phosphatase', 'transferase']
TIER3_CLASSES = ['transcription factor', 'adaptor', 'scaffold', 'structural']
def assess_druggability(genes, include_known_drugs=True, include_protein_class=True,
include_ppi_inhibitors=False):
"""
Assess druggability of gene targets.
Parameters
----------
genes : list
List of gene symbols
include_known_drugs : bool
Include known drug information from DrugBank/ChEMBL (default: True)
include_protein_class : bool
Include protein class annotations (default: True)
include_ppi_inhibitors : bool
Consider protein-protein interaction inhibitors (default: False)
Returns
-------
pandas.DataFrame
Druggability scores and annotations
"""
print(f"Assessing druggability for {len(genes)} genes...")
results = []
for gene in genes:
# Get protein class (placeholder - would query UniProt/InterPro)
protein_class = get_protein_class(gene)
# Determine druggability tier
druggability_tier = classify_druggability(protein_class, include_ppi_inhibitors)
# Get known drugs (placeholder - would query DrugBank/ChEMBL)
known_drugs = get_known_drugs(gene) if include_known_drugs else []
# Calculate druggability score (0-1)
druggability_score = calculate_druggability_score(
druggability_tier,
len(known_drugs),
protein_class
)
# Check for clinical precedent
clinical_precedent = len(known_drugs) > 0
results.append({
'gene': gene,
'protein_class': protein_class,
'druggability_tier': druggability_tier,
'druggability_score': druggability_score,
'known_drugs': '; '.join(known_drugs) if known_drugs else 'None',
'n_known_drugs': len(known_drugs),
'clinical_precedent': clinical_precedent
})
druggability_df = pd.DataFrame(results)
print(f"\nDruggability summary:")
print(f" Tier 1 (High): {(druggability_df['druggability_tier'] == 'Tier 1').sum()}")
print(f" Tier 2 (Medium): {(druggability_df['druggability_tier'] == 'Tier 2').sum()}")
print(f" Tier 3 (Low): {(druggability_df['druggability_tier'] == 'Tier 3').sum()}")
print(f" Tier 4 (Very Low): {(druggability_df['druggability_tier'] == 'Tier 4').sum()}")
print(f" Genes with known drugs: {druggability_df['clinical_precedent'].sum()}")
scripts/export_results.py
"""
TWAS Results Export and Reporting
This module provides functions for generating comprehensive target reports
with genetic evidence, directionality, causality, and druggability.
"""
import pandas as pd
from pathlib import Path
def generate_target_report(twas_results, coloc_results, therapeutic_df,
mr_results=None, druggability_scores=None,
output_file="reports/target_report.xlsx"):
"""
Generate comprehensive Excel report for biopharma target prioritization.
Parameters
----------
twas_results : pandas.DataFrame
Full TWAS results
coloc_results : pandas.DataFrame
Colocalization results
therapeutic_df : pandas.DataFrame
Therapeutic directionality recommendations
mr_results : pandas.DataFrame, optional
Mendelian Randomization results
druggability_scores : pandas.DataFrame, optional
Druggability assessment
output_file : str
Output Excel file path
Returns
-------
str
Path to generated report
"""
output_path = Path(output_file)
output_path.parent.mkdir(parents=True, exist_ok=True)
print(f"Generating comprehensive target report...")
# Create Excel writer
with pd.ExcelWriter(output_file, engine='openpyxl') as writer:
# Sheet 1: Executive Summary
executive_summary = create_executive_summary(
therapeutic_df, coloc_results, druggability_scores, mr_results
)
executive_summary.to_excel(writer, sheet_name='Executive Summary', index=False)
print(" ✓ Executive Summary")
# Sheet 2: Full TWAS Results
twas_results.to_excel(writer, sheet_name='TWAS Results', index=False)
print(" ✓ TWAS Results")
# Sheet 3: Colocalization Details
coloc_results.to_excel(writer, sheet_name='Colocalization', index=False)
print(" ✓ Colocalization")
# Sheet 4: Therapeutic Directionality
therapeutic_df.to_excel(writer, sheet_name='Directionality', index=False)
print(" ✓ Therapeutic Directionality")
# Sheet 5: Mendelian Randomization (if available)
if mr_results is not None and not mr_results.empty:
mr_results.to_excel(writer, sheet_name='Mendelian Randomization', index=False)
print(" ✓ Mendelian Randomization")
# Sheet 6: Druggability Assessment (if available)
if druggability_scores is not None and not druggability_scores.empty:
druggability_scores.to_excel(writer, sheet_name='Druggability', index=False)
print(" ✓ Druggability")
print(f"\nReport saved: {output_file}")
return str(output_file)
def create_executive_summary(therapeutic_df, coloc_results, druggability_scores, mr_results):
"""