PCR Primer Design
Design and validate primers for PCR, qPCR and sequencing.
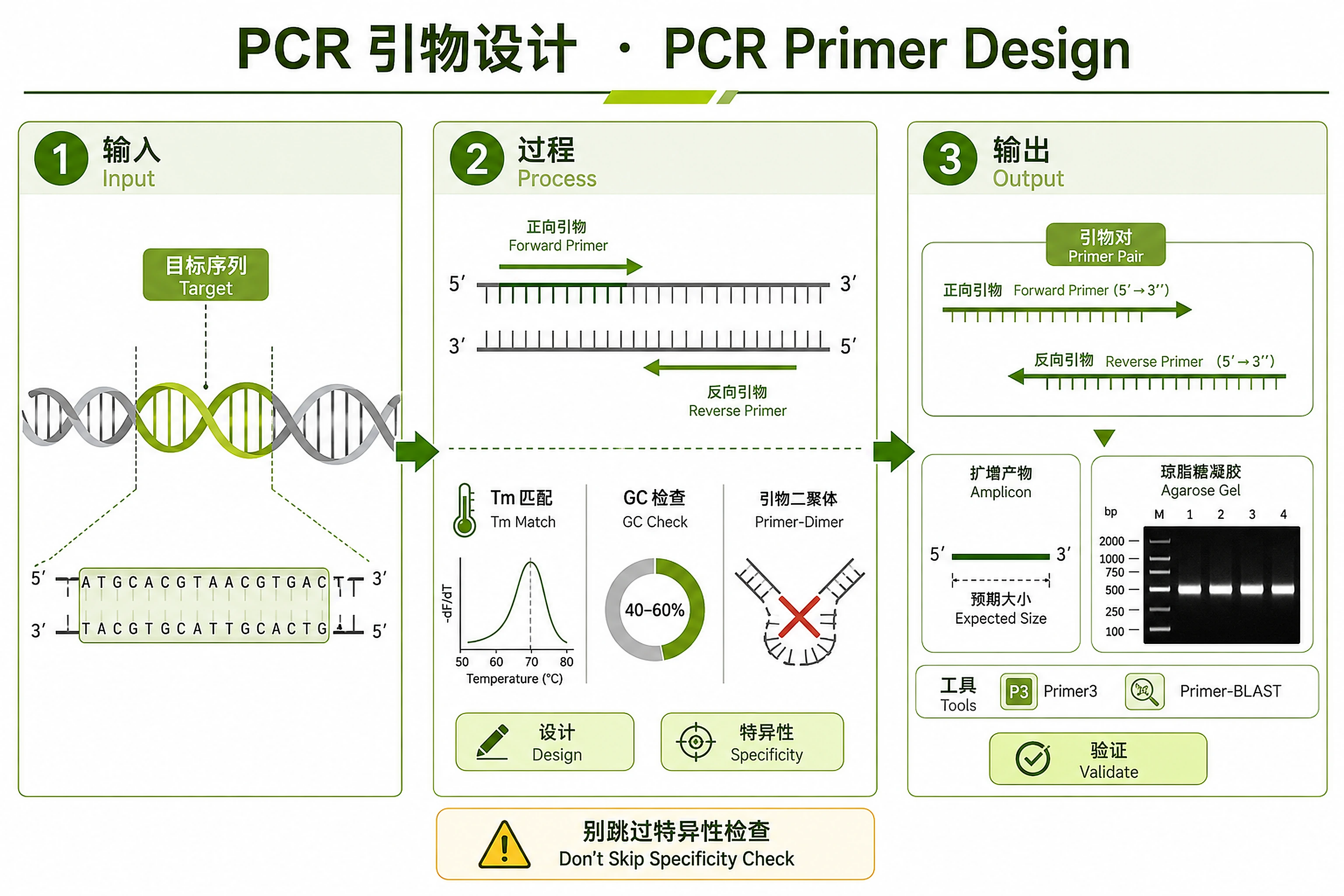
Overview
Problem. Need specific, dimer-free, valid primers to amplify DNA.
Learning goals
- Hard constraints: Tm match, no dimers, specificity
- Requirements differ by application
Figures
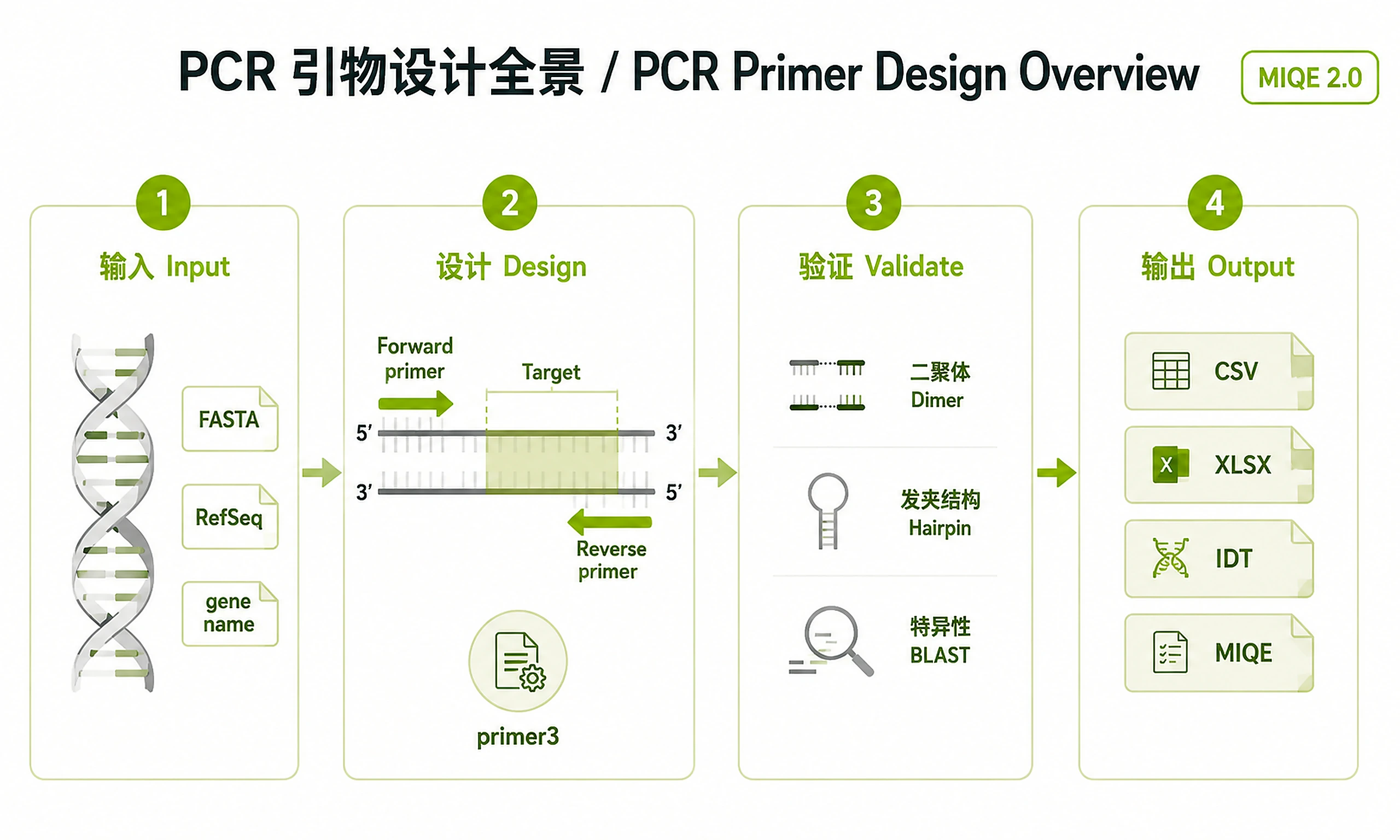

Tutorial
Comprehensive PCR and qPCR primer design following MIQE 2.0 guidelines with automated validation.
When to Use This Skill
Use this skill when you need:
- ✅ qPCR primers with MIQE 2.0 compliance for publication
- ✅ Standard PCR primers for cloning, genotyping, or amplification (100-1000 bp)
- ✅ TaqMan probes for probe-based qPCR assays
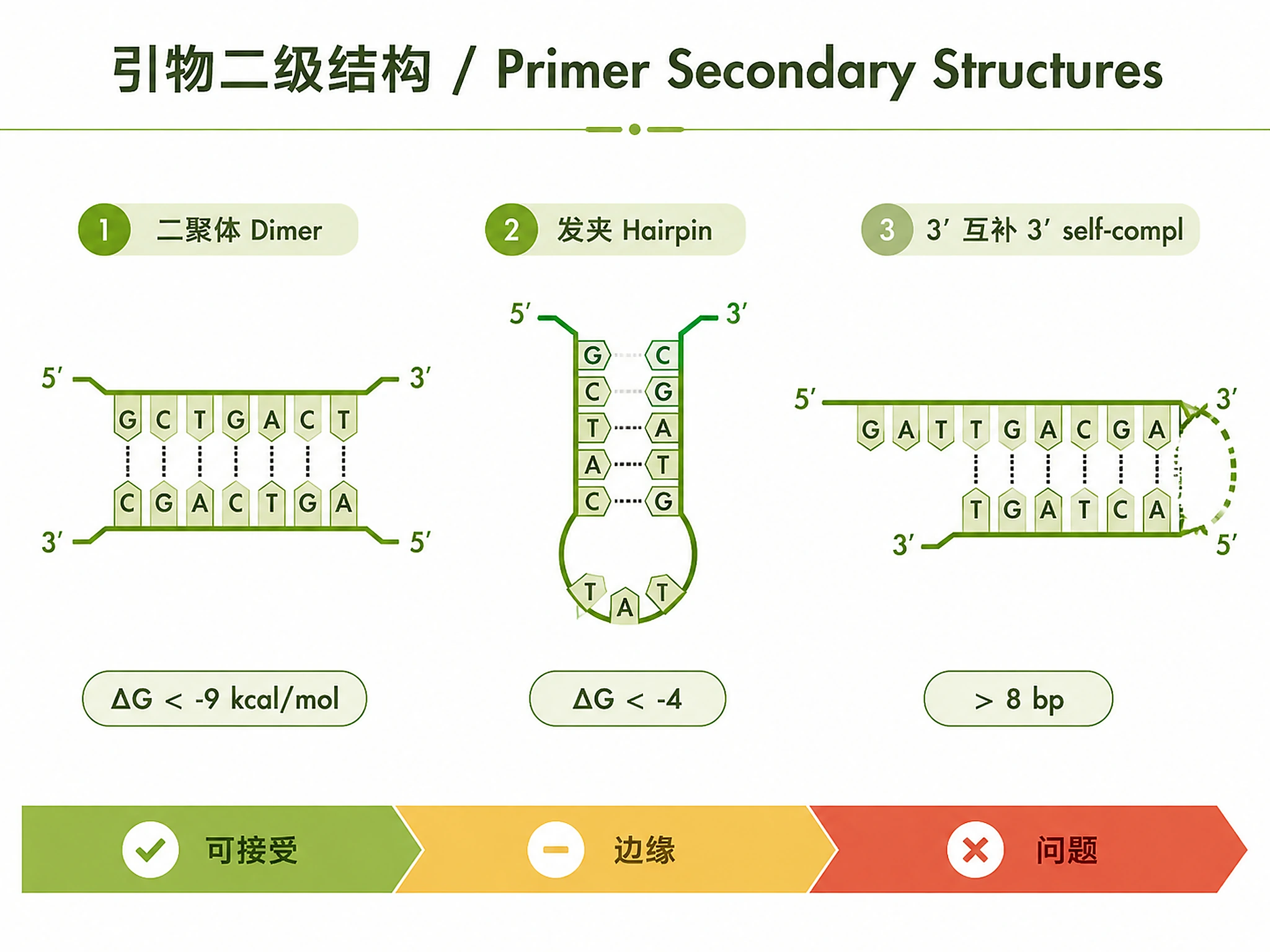
- ✅ Rigorous validation (specificity, dimers, secondary structures)
- ✅ Publication-quality documentation with comprehensive reports
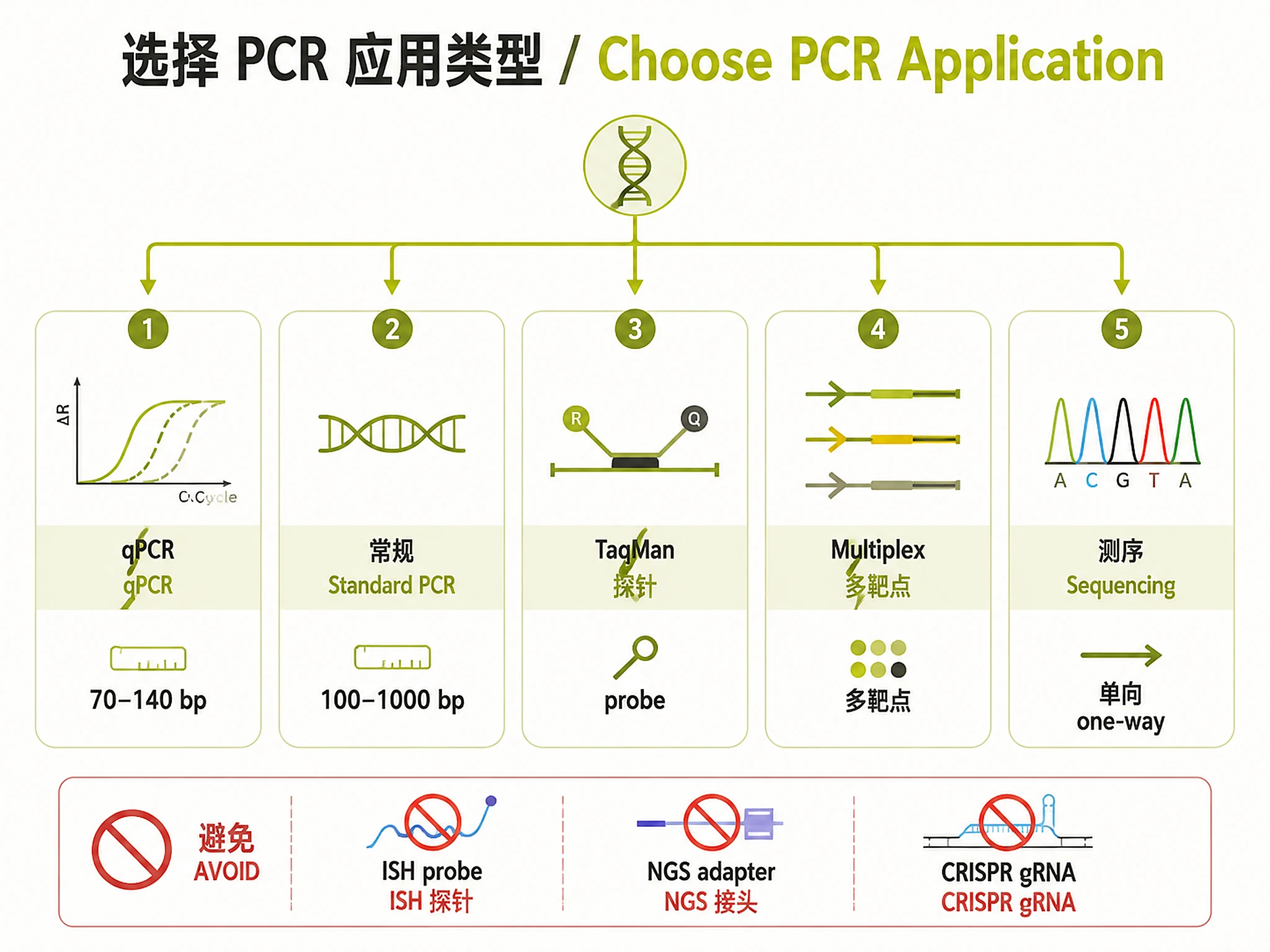
Choose application based on:
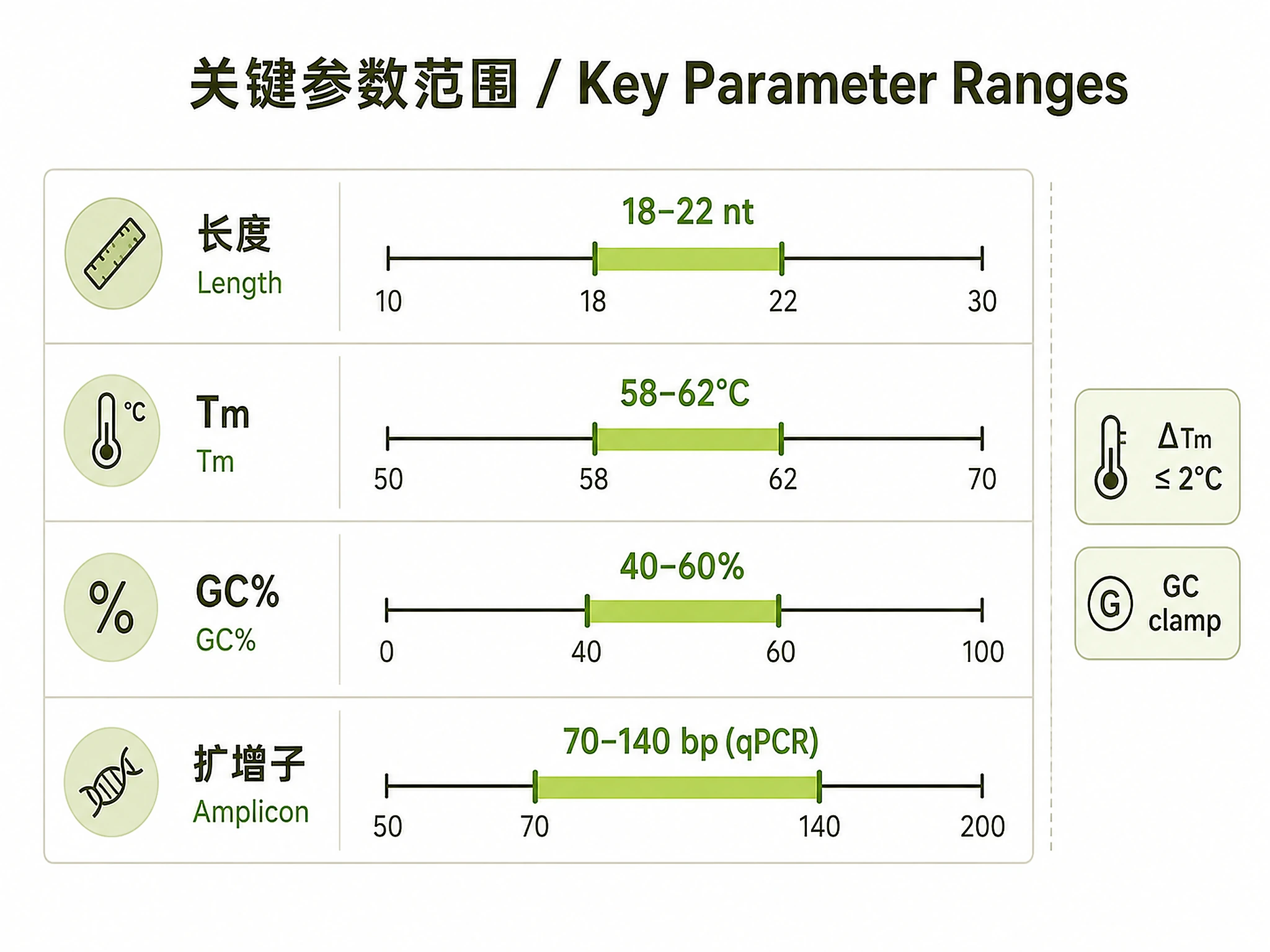
- qPCR: 70-140 bp amplicons, strict Tm matching (±2°C), MIQE compliance
- Standard PCR: 100-1000 bp amplicons, general amplification
- TaqMan: Probe-based detection, fluorescent assays
- Multiplex: Multiple targets, compatible Tm requirements
- Sequencing: Single-direction primers, Sanger sequencing
Don't use for:
- ❌ In-situ hybridization probes → use specialized oligo design tools
- ❌ NGS library prep primers → use adapter design workflows
- ❌ CRISPR guide RNAs → use CRISPR-specific design tools
Quick Start (Example)
Test this skill with a sample qPCR design in ~2 minutes:
# Example: Design qPCR primers for a 700bp target sequence
from scripts.design_qpcr_primers import design_qpcr_primers
# Sample GAPDH sequence (700 bp, exon 3-4 region)
sequence = "ATGGGGAAGGTGAAGGTCGGAGTCAACGGATTTGGTCGTATTGGGCGCCTGGTCACCAGGGCTGCTTTTAACTCTGGTAAAGTGGATATTGTTGCCATCAATGACCCCTTCATTGACCTCAACTACATGGTTTACATGTTCCAATATGATTCCACCCATGGCAAATTCCATGGCACCGTCAAGGCTGAGAACGGGAAGCTTGTCATCAATGGAAATCCCATCACCATCTTCCAGGAGCGAGATCCCTCCAAAATCAAGTGGGGCGATGCTGGCGCTGAGTACGTCGTGGAGTCCACTGGCGTCTTCACCACCATGGAGAAGGCTGGGGCTCATTTGCAGGGGGGAGCCAAAAGGGTCATCATCTCTGCCCCCTCTGCTGATGCCCCCATGTTCGTCATGGGTGTGAACCATGAGAAGTATGACAACAGCCTCAAGATCATCAGCAATGCCTCCTGCACCACCAACTGCTTAGCACCCCTGGCCAAGGTCATCCATGACAACTTTGGTATCGTGGAAGGACTCATGACCACAGTCCATGCCATCACTGCCACCCAGAAGACTGTGGATGGCCCCTCCGGGAAACTGTGGCGTGATGGCCGCGGGGCTCTCCAGAACATCATCCCTGCCTCTACTGGCGCTGCCAAGGCTGTGGGCAAGGTCATCCCTGAGCTGAACGGGAAGCTCACTGGCATGGCCTTCCGTGTCCCCACTGCCAACGTGTCAGTGGTGGACCTGACCTGCCGTCTAGAAAAACCTGCCAAATATGATGACATCAAGAAGGTGGTGAAGCAGGCGTCGGAGGGCCCCCTCAAGGGCATCCTGGGCTACACTGAGCACCAGGTGGTCTCCTCTGACTTCAACAGCGACACCCACTCCTCCACCTTTGACGCTGGGGCTGGCATTGCCCTCAACGACCACTTTGTCAAGCTCATTTCCTGGTATGACAACGAATTTGGCTACAGCAACAGGGTGGTGGACCTCATGGCCCACATGGCCTCCAAGGAGTAAGACCCCTGGACCACCAGCCCCAGCAAGAGCACAAGAGGAAGAGAGAGACCCTCACTGCTGGGGAGTCCCTGCCACACTCAGTCCCCCACCACACTGAATCTCCCCTCCTCACAGTTGCCATGTAGACCCCTTGAAGAGGGGAGGGCTCTCTCTTCCTCTTGTGCTCTTGCTGGGGCTGGCATTGCCCTCAACGACCACTTTGTCAAGCTCATTTCCTGGTATGACAACG"
primers = design_qpcr_primers(
sequence=sequence,
amplicon_size_range=(80, 120),
num_return=5
)
print(f"Found {len(primers['primers'])} primer pairs")
print(f"MIQE-compliant: {sum(p.get('miqe_compliant', False) for p in primers['primers'])}")
What you get: 3-5 MIQE-compliant primer pairs, amplicons 80-120 bp, Tm matched within 2°C
For your own data: Follow Clarification Questions below to provide your sequence.
Installation
Core packages:
pip install primer3-py biopython plotnine plotnine-prism pandas requests openpyxl
Recommended: Use virtual environment:
python -m venv pcr_env
source pcr_env/bin/activate # Windows: pcr_env\Scripts\activate
pip install -r requirements.txt
Software Requirements
| Software | Version | License | Commercial Use | Installation |
|---|---|---|---|---|
| primer3-py | ≥2.0.0 | GPL v2 | ✅ Permitted* | pip install primer3-py |
| Biopython | ≥1.80 | BSD | ✅ Permitted | pip install biopython |
| plotnine | ≥0.12.0 | MIT | ✅ Permitted | pip install plotnine |
| plotnine-prism | latest | MIT | ✅ Permitted | pip install plotnine-prism |
| pandas | ≥1.5.0 | BSD | ✅ Permitted | pip install pandas |
*GPL v2 permits use in AI agent applications (execution, not distribution).
NCBI API: Primer-BLAST access is free. Rate limit: 3 requests/second (no API key) or 10/second (with free API key). See references/primer_design_best_practices.md#ncbi-api-setup for API key setup.
Inputs
Required:
- Target DNA sequence in one of these formats:
- FASTA file (local or uploaded)
- GenBank/RefSeq accession (e.g., NM_002046)
- Raw sequence (paste directly)
- Gene name + organism (fetches from NCBI)
Sequence requirements:
- Minimum length: 150 bp (qPCR), 300 bp (standard PCR)
- Format: ATCG nucleotides (U converted to T)
- Quality: Avoid ambiguous bases (N) in primer regions
Optional:
- Regions to avoid (SNPs, repeats, splice sites)
- Custom parameter ranges (Tm, GC%, amplicon size)
- Organism/genome for specificity checking
See references/primer_design_best_practices.md#input-preparation for sequence preparation guidelines.
Outputs
Primary results:
- Primer sequences with properties (Tm, GC%, length, position)
- Validation report (dimers, secondary structures, specificity)
- Quality scores and QC flags
Export formats (user-selectable):
primers.csv- Spreadsheet-compatible tableprimers.xlsx- Excel with multiple sheets (design, validation, parameters)primers.json- Structured data for programmatic useidt_order.txt- IDT ordering format (copy-paste ready)miqe_checklist.xlsx- MIQE 2.0 compliance documentation (qPCR only)
Visualizations (optional):
- Primer binding site alignment (SVG, 300 DPI)
- Tm distribution plots
- Secondary structure diagrams
See references/code_examples.md#export-examples for format details.
Clarification Questions
1. Input Sequence (ASK THIS FIRST)
Do you have a specific DNA sequence to design primers for?
- Option A: Upload FASTA file or provide file path
- Option B: Provide GenBank/RefSeq accession (e.g., NM_001256799)
- Option C: Provide gene name + organism (will fetch from NCBI)
- Option D: Paste sequence directly
If uploaded: Is this the complete target sequence or a specific region?
Expected: 150+ bp for qPCR, 300+ bp for standard PCR
2. PCR Application
What is your intended use for these primers?
- qPCR (Quantitative PCR) - Gene expression, 70-140 bp amplicons, MIQE-compliant
- Standard PCR - General amplification, cloning, genotyping, 100-1000 bp amplicons
- TaqMan Assay - Probe-based qPCR with fluorescent detection
- Multiplex PCR - Multiple targets simultaneously, compatible Tm required
- Sequencing - Sanger sequencing, single-direction primer
- SNP Genotyping - Allele-specific amplification
Default: qPCR (most common for gene expression studies)
3. Design Parameters
Do you want to use application-specific default parameters or customize?
- Standard parameters (recommended) - Optimized for selected application
- Custom Tm range - Specify melting temperature range (default: 58-62°C for qPCR)
- Custom amplicon size - Specify product size range
- Custom GC range - Adjust GC% (default: 40-60%)
- Avoid regions - Exclude specific sequences (SNPs, repeats, etc.)
For qPCR: Target exon-exon junction or ensure intron >1kb (MIQE guideline)?
To understand design parameters: See references/parameter_ranges.md
4. Validation Level
How thoroughly should primers be validated?
- Basic - Tm, GC%, dimer check (~1 min, sufficient for most uses)
- Standard - Basic + in-silico PCR (~2-3 min, recommended)
- Complete - Standard + NCBI Primer-BLAST (~5-10 min, publication-quality)
- MIQE-compliant - Complete + full documentation (qPCR only, ~10 min)
Note: NCBI Primer-BLAST requires internet and respects rate limits (slower but most thorough).
5. Output Requirements
What outputs do you need?
- Export format: CSV (default), Excel, JSON, IDT order format, or MIQE checklist
- Report format: Markdown (default), HTML, or text
- Visualizations: Generate primer alignment plots? (yes/no)
- Number of primers: How many primer pairs to return? (default: 5)
For qPCR: MIQE checklist is automatically generated.
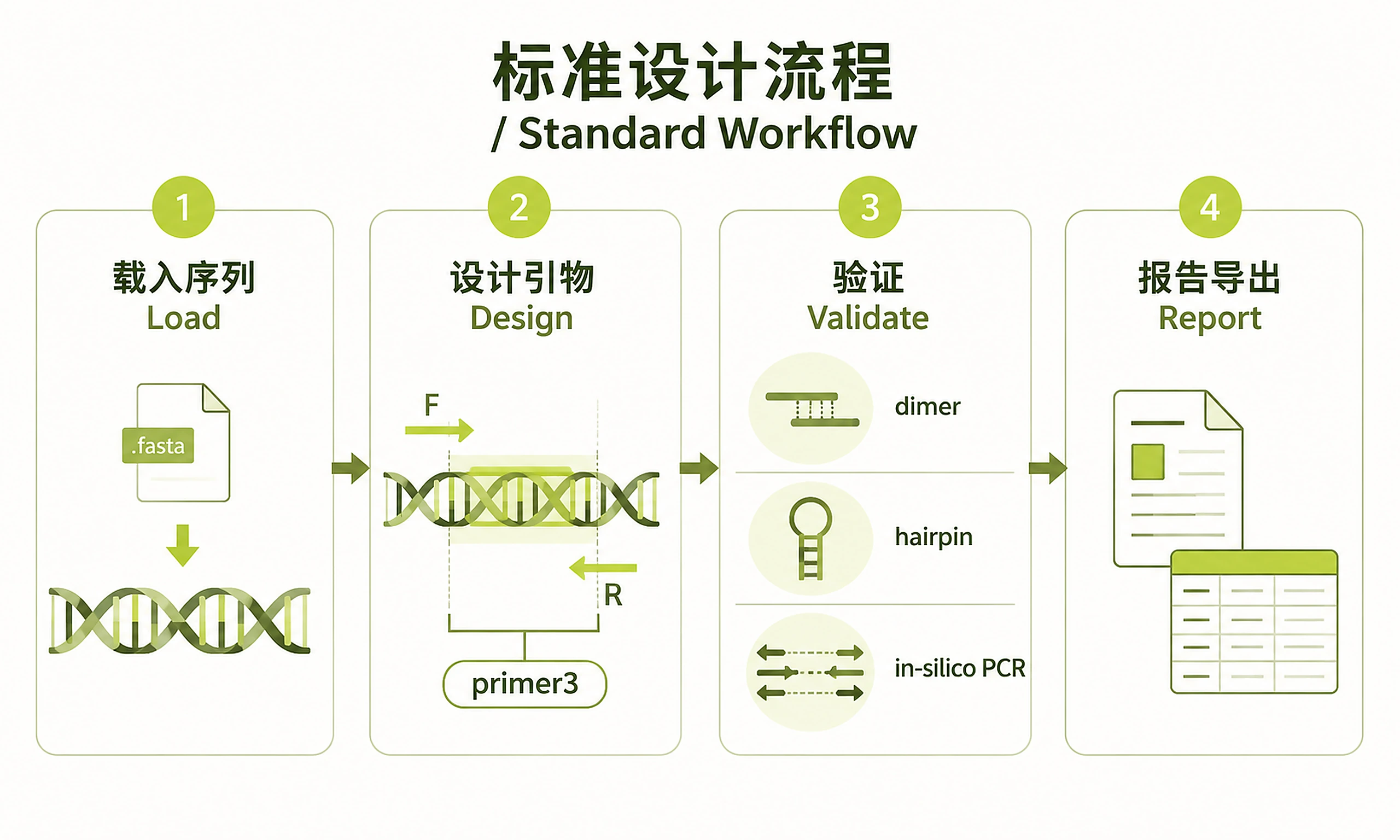
Standard Workflow
🚨 EXECUTE EXACTLY AS SHOWN - Do not modify these commands.
CRITICAL: Use relative paths (scripts/, references/). DO NOT construct absolute paths.
Step 1: Load Target Sequence
Option A: Load from FASTA file
from Bio import SeqIO
record = SeqIO.read("your_sequence.fasta", "fasta")
sequence = str(record.seq)
Option B: Load from GenBank accession
See references/code_examples.md#loading-sequences for NCBI fetching code.
Option C: Paste sequence directly
# For quick testing, paste your target sequence
sequence = "ATGGGGAAGGTGAAGGTCGGAGTCAACGGATTTGGTCGTATTGGG..." # Your sequence here
Step 2: Design Primers
Choose based on application (from Clarification Question #2):
For qPCR (most common):
from scripts.design_qpcr_primers import design_qpcr_primers
primers = design_qpcr_primers(
sequence=sequence,
amplicon_size_range=(70, 140), # MIQE guideline
tm_match_threshold=2.0,
num_return=5
)
For Standard PCR:
from scripts.design_standard_primers import design_pcr_primers
primers = design_pcr_primers(
sequence=sequence,
amplicon_size_range=(100, 1000),
tm_range=(55, 65),
num_return=5
)
For TaqMan Assay:
from scripts.design_taqman_probes import design_taqman_assay
assay = design_taqman_assay(
sequence=sequence,
probe_tm_offset=8.0, # Probe Tm = primer Tm + 8°C
num_return=5
)
For custom parameters: Read references/parameter_ranges.md and adapt ranges to your requirements.
Step 3: Validate Primers
Basic validation (recommended for all):
from scripts.check_dimers import analyze_dimers
from scripts.check_secondary_structures import analyze_secondary_structures
top_primer = primers['primers'][0]
# Check dimers
dimer_result = analyze_dimers(
[top_primer['forward_seq'], top_primer['reverse_seq']],
temperature=60.0
)
# Check secondary structures
fwd_structure = analyze_secondary_structures(top_primer['forward_seq'], 60.0)
rev_structure = analyze_secondary_structures(top_primer['reverse_seq'], 60.0)
Complete validation (for publication):
from scripts.validate_specificity import in_silico_pcr
# In-silico PCR (fast)
products = in_silico_pcr(
forward_primer=top_primer['forward_seq'],
reverse_primer=top_primer['reverse_seq'],
sequence=sequence
)
# For NCBI Primer-BLAST: See [references/code_examples.md#validation-examples](references/code_examples.md#validation-examples)
# Note: Requires internet, respects rate limits (3 req/sec)
⚠️ CRITICAL - DO NOT:
- ❌ Use absolute paths like
/mnt/knowhow/→ use relative pathsscripts/ - ❌ Write inline primer design code → use provided scripts
- ❌ Skip dimer/structure checks → these catch common failures
Step 4: Generate Reports and Export
from scripts.generate_reports import generate_primer_report
from scripts.export_results import export_primers
# Generate report
report = generate_primer_report(
primers=primers,
validation_results={
'dimers': dimer_result,
'secondary_structures': {'forward': fwd_structure, 'reverse': rev_structure}
},
output_format="markdown",
include_miqe_checklist=(application == 'qpcr') # From Clarification Question #2
)
# Export in requested format(s)
export_primers(primers, format="csv", output_file="primers.csv")
export_primers(primers, format="excel", output_file="primers.xlsx")
# For qPCR: MIQE checklist
if application == 'qpcr':
export_primers(primers, format="miqe_checklist", output_file="miqe_checklist.xlsx")
For visualization plots: See references/code_examples.md#visualization
That's it! The scripts handle all design and validation automatically.
Common Issues
| Issue | Likely Cause | Solution |
|---|---|---|
| No primers found | Target too short or constraints too strict | Relax Tm range (±5°C), widen GC range (35-65%), provide longer sequence (≥300 bp) |
| High primer-dimer formation | Complementary sequences, low Tm | Increase Tm to 60-62°C, redesign avoiding complementary regions |
| Multiple off-target amplicons | Low specificity, repetitive sequences | Move to unique region, increase primer length (22-25 nt), check specificity with BLAST |
| Tm mismatch between primers | Different GC content | Adjust primer lengths to balance Tm, use Primer3 penalty weights (see references/parameter_ranges.md) |
| Poor qPCR efficiency | Dimers, secondary structures, amplicon >150 bp | Redesign with shorter amplicon (70-120 bp), check for hairpins, verify no dimers |
| NCBI API rate limit errors | Too many requests too quickly | Wait 0.33 sec between requests, get free API key (10 req/sec), or use in-silico PCR first |
| ImportError for scripts | Missing __init__.py in scripts/ |
Create empty scripts/__init__.py file to make it a Python package |
For detailed troubleshooting: See references/troubleshooting_guide.md
Suggested Next Steps
After successful primer design:
- Order primers - Use IDT order format export for direct ordering
- Optimize PCR conditions - Test annealing temperature gradient (Tm ± 3°C)
- Validate experimentally: - Test specificity (gel electrophoresis, melt curve for qPCR) - Optimize primer concentration (50-900 nM range) - Verify amplicon size (gel or bioanalyzer)
- For qPCR - Perform standard curve, efficiency calculation, melt curve analysis
- Document - Save MIQE checklist and validation reports for publication
Related protocols: See references/primer_design_best_practices.md#experimental-validation
Related Skills
- qPCR Data Analysis - Analyze qPCR Cq values after experimental validation
- Sanger Sequencing Analysis - Analyze results from sequencing primers
- Gene Expression Normalization - Choose reference genes for qPCR
References
Documentation
- Primer Design Best Practices - Comprehensive design guidelines
- MIQE 2.0 Guidelines - qPCR standards and compliance
- Parameter Ranges - Recommended parameter values
- Troubleshooting Guide - Common issues and solutions
- Code Examples - Complete code examples for all applications
Key Publications
- MIQE 2.0: Bustin SA, et al. (2025) Clinical Chemistry 71(6):634-660
- Primer3: Untergasser A, et al. (2012) Nucleic Acids Research 40(15):e115
- Primer-BLAST: Ye J, et al. (2012) BMC Bioinformatics 13:134
Online Resources
- NCBI Primer-BLAST: https://www.ncbi.nlm.nih.gov/tools/primer-blast/
- Primer3 Web: https://primer3.ut.ee/
- MIQE Guidelines: https://rdml.org/miqe.html
Code preview
scripts/__init__.py
# This file makes the scripts directory a Python package
# Required for imports like: from scripts.design_qpcr_primers import design_qpcr_primersscripts/calculate_tm.py
"""
Calculate melting temperatures using multiple methods.
This module provides functions for calculating primer Tm using various
algorithms and salt correction methods.
"""
import primer3
from Bio.SeqUtils import MeltingTemp as mt
from Bio.Seq import Seq
from typing import Dict
def calculate_tm_comprehensive(
primer_sequence: str,
method: str = "nearest_neighbor",
salt_conc: float = 50.0,
dna_conc: float = 200.0,
mg_conc: float = 0.0,
dntp_conc: float = 0.0
) -> Dict:
"""
Calculate melting temperature using multiple methods.
Parameters
----------
primer_sequence : str
Primer sequence
method : str
Tm calculation method:
- "nearest_neighbor" (most accurate, default)
- "salt_adjusted" (salt-adjusted formula)
- "gc_content" (simple GC% method)
- "all" (calculate using all methods)
salt_conc : float
Monovalent cation (Na+, K+) concentration in mM. Default: 50.0
dna_conc : float
DNA/primer concentration in nM. Default: 200.0
mg_conc : float
Mg2+ concentration in mM. Default: 0.0
dntp_conc : float
dNTP concentration in mM. Default: 0.0
Returns
-------
dict
Dictionary with Tm values from different methods
Example
-------
>>> tm_results = calculate_tm_comprehensive(
... "ATGCGTACGATCGATCG",
... method="all",
... salt_conc=50.0
... )
>>> print(f"Nearest-Neighbor Tm: {tm_results['tm_nn']:.1f}°C")
"""
# Validate input
primer_sequence = primer_sequence.upper().strip()
if not all(base in 'ATCGN' for base in primer_sequence):
raise ValueError("Primer must contain only A, T, C, G, or N")
if len(primer_sequence) < 10:
raise ValueError("Primer too short for accurate Tm calculation (minimum 10 bp)")
results = {
'primer_sequence': primer_sequence,
'primer_length': len(primer_sequence),
}
# Method 1: Nearest-Neighbor (most accurate)
if method in ["nearest_neighbor", "all"]:
# Using primer3
tm_nn_primer3 = primer3.calcTm(
primer_sequence,
mv_conc=salt_conc,
dv_conc=mg_conc,
dntp_conc=dntp_conc,scripts/check_dimers.py
"""
Check for primer dimers and cross-dimers.
This module analyzes primer-primer interactions to identify potential
dimer formation that can reduce PCR efficiency.
"""
import primer3
from typing import List, Dict
def analyze_dimers(
primer_list: List[str],
temperature: float = 60.0,
dg_threshold: float = -5.0,
mv_conc: float = 50.0,
dv_conc: float = 0.0,
dntp_conc: float = 0.8,
dna_conc: float = 50.0
) -> Dict:
"""
Analyze primer dimers and cross-dimers for a set of primers.
Parameters
----------
primer_list : list of str
List of primer sequences to analyze
temperature : float
PCR annealing temperature in °C. Default: 60.0
dg_threshold : float
ΔG threshold in kcal/mol for flagging dimers. Default: -5.0
Dimers with ΔG < threshold are likely to form
mv_conc : float
Monovalent cation concentration in mM. Default: 50.0
dv_conc : float
Divalent cation concentration in mM. Default: 0.0
dntp_conc : float
dNTP concentration in mM. Default: 0.8
dna_conc : float
DNA concentration in nM. Default: 50.0
Returns
-------
dict
Dictionary containing:
- 'interactions': list of all primer-primer interactions
- 'problematic_dimers': list of dimers below threshold
- 'has_issues': bool indicating if any dimers exceed threshold
Example
-------
>>> primers = ["ATGCGTACGATCGATCG", "CGTAGCTAGCTAGCTAG"]
>>> analysis = analyze_dimers(primers, temperature=60.0)
>>> if analysis['has_issues']:
... print("⚠ Primer dimers detected!")
"""
# Validate inputs
primer_list = [p.upper().strip() for p in primer_list]
if len(primer_list) < 2:
raise ValueError("Need at least 2 primers to check for dimers")
for primer in primer_list:
if not all(base in 'ATCGN' for base in primer):
raise ValueError(f"Invalid primer sequence: {primer}")
interactions = []
problematic_dimers = []
# Check all pairwise interactions
for i, primer1 in enumerate(primer_list):
for j, primer2 in enumerate(primer_list):
if i <= j: # Check each pair once, including self-dimers
# Determine interaction type
if i == j:
interaction_type = "self-dimer"
primer1_name = f"Primer_{i+1}"
primer2_name = f"Primer_{i+1}"
else:Companion files
| Type | Path | Bytes |
|---|---|---|
| Markdown | references/code_examples.md | 14,135 |
| Markdown | references/miqe_guidelines.md | 11,475 |
| Markdown | references/parameter_ranges.md | 9,526 |
| Markdown | references/primer_design_best_practices.md | 11,697 |
| Markdown | references/troubleshooting_guide.md | 14,013 |
| Python | scripts/__init__.py | 146 |
| Python | scripts/calculate_tm.py | 9,719 |
| Python | scripts/check_dimers.py | 9,356 |
| Python | scripts/check_secondary_structures.py | 10,008 |
| Python | scripts/design_qpcr_primers.py | 8,249 |
| Python | scripts/design_standard_primers.py | 6,683 |
| Python | scripts/design_taqman_probes.py | 7,678 |
| Python | scripts/export_results.py | 12,444 |
| Python | scripts/generate_reports.py | 13,653 |
| Python | scripts/validate_specificity.py | 9,306 |
| Python | scripts/visualize_primers.py | 10,582 |
| Markdown | SKILL.md | 14,815 |
| JSON | skill.meta.json | 3,302 |