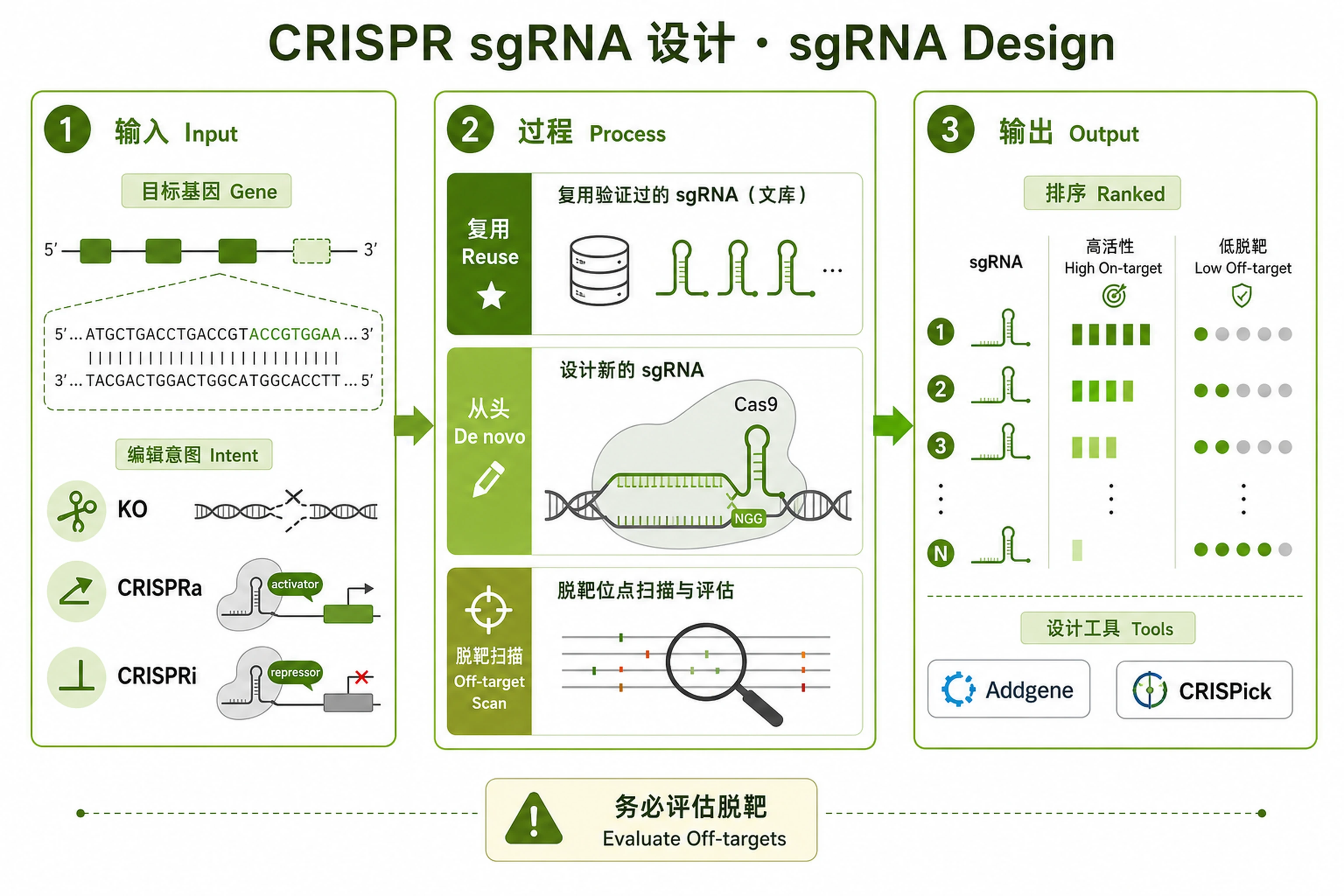
sgRNA Design
Find or design guides — validated-first, then de novo.
Overview
Problem. Need efficient, low-off-target guides to KO/activate/inhibit.
Learning goals
- Reuse validated guides before designing de novo
- Balance on-target activity and off-target risk
Figures

Tutorial
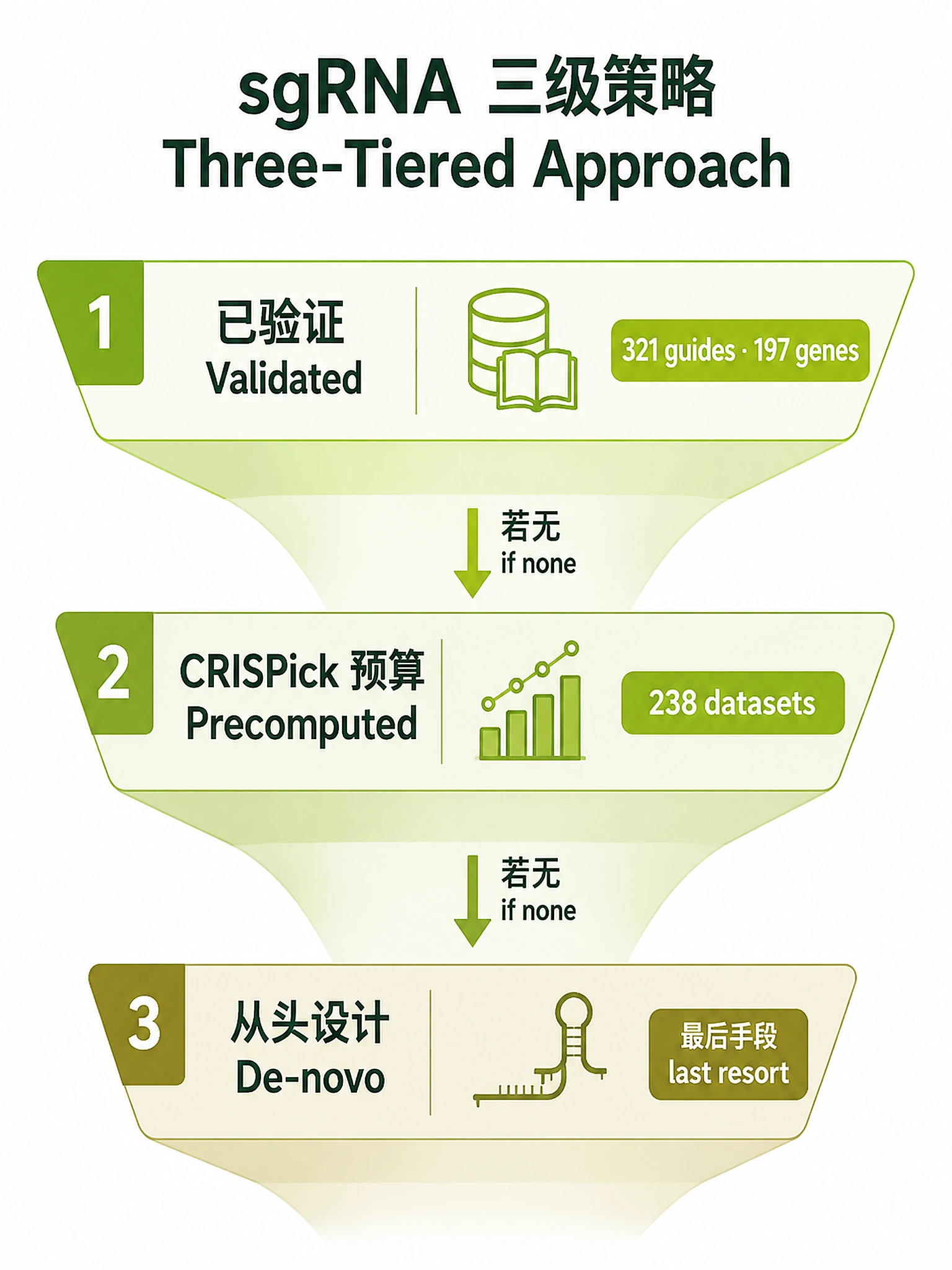
Find or design sgRNAs by prioritizing validated sequences before computational predictions.
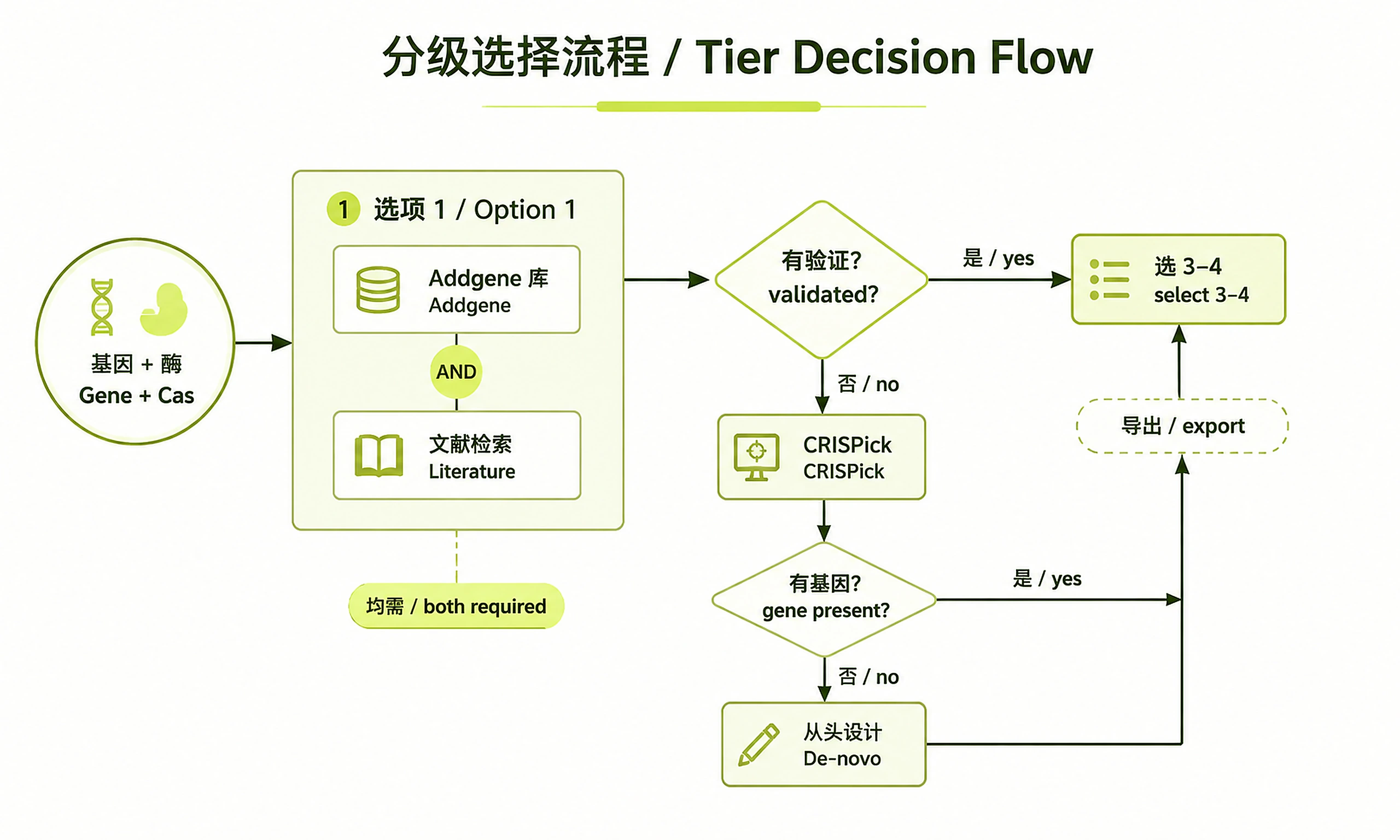
Always start at Option 1 and only descend to the next tier when the current one yields nothing
usable. Ported from the Biomni sgRNA_design_guide.md (snap-stanford/Biomni), with the data
parsing corrected and the literature step wired to this environment's search tools.
When to use
- "Give me an sgRNA to knock out TP53 in human cells"
- "Design CRISPR guides to activate OCT4" / "CRISPRi guides for MYC"
- "What guide RNA should I use for
with SpCas9 / SaCas9 / Cas12a?" - Selecting guides for an arrayed or pooled CRISPR screen
Inputs
- Gene symbol (required), e.g. TP53, BRCA1, AAVS1.
- Organism (default human), e.g. human/mouse/rat or NCBI TAXID.
- Application (default knockout): knockout / activation (CRISPRa) / inhibition (CRISPRi).
- Cas enzyme (default SpCas9): SpCas9, SaCas9, AsCas12a, enAsCas12a.
Outputs
<GENE>_selected_sgRNAs.csv— unified table of 3–4 recommended guides (sequence, source, rank/score, exon/position, PAM, citation/dataset, notes).<GENE>_sgRNA_summary.md— which tier was used and why, the picks, and caveats.
Save both to the user's results directory.
Bundled resources (work offline; refresh via references/refresh_resources.md)
references/resource/addgene_grna_sequences.csv— 321 validated sgRNAs, 197 genes (Addgene).references/resource/CRISPick_download_links.txt— 238 CRISPick dataset URLs, 13 organisms.
Scripts
| Script | Purpose |
|---|---|
scripts/search_addgene.py |
Tier 1 / Method 1 — search the Addgene database (handles the HTML-wrapped IDs and messy species values). |
scripts/find_crispick_dataset.py |
Tier 2 / Step 1 — resolve the correct CRISPick download URL for organism+enzyme+application. |
scripts/select_crispick_sgrnas.py |
Tier 2 / Step 3 — filter a downloaded CRISPick file to your gene, rank, pick 3–4. |
scripts/check_design_rules.py |
Tier 3 — sanity-check a candidate guide (length, GC, TTTT, PAM). |
scripts/export_results.py |
Write the unified CSV + markdown summary. |
Option 1 — Validated sequences (ALWAYS try first)
You MUST complete both Method 1 and Method 2 before considering Option 2. Do not skip Method 2 even if Method 1 finds nothing — many validated guides live only in the literature.
Method 1 — Bundled Addgene database
import sys; sys.path.insert(0, "scripts")
from search_addgene import search_addgene
hits = search_addgene("TP53", species="human", application="knockout")
print(len(hits)) # 0 for TP53 -> still do Method 2 before Option 2
Each hit carries a clean Target Sequence, pubmed_id/pubmed_url, plasmid_id/plasmid_url,
and Depositor. Cite the PubMed ID of the original publication in your methods.
application accepts intent words and maps them to Addgene's vocabulary:
knockout→cut, activation→activate, inhibition/CRISPRi→interfere/RNA targeting.
Method 2 — Literature & web search (REQUIRED)
This environment does not expose advanced_web_search_claude; use the available tools instead.
Run both for coverage:
LiteratureSearch— peer-reviewed papers (validated guides, supplements).WebSearch— vendor/database hits (GenScript, Horizon, lab protocols).
Query templates (substitute the gene):
"sgRNA" OR "guide RNA" "<GENE>" validated experimental
"CRISPR knockout" "<GENE>" sgRNA sequence validated
"<GENE>" sgRNA "cutting efficiency" OR "on-target"
Scan ≥10–15 results and check supplementary materials. For any validated guide, record the sequence, citation (PMID/DOI), and validation details (cell line, cutting efficiency).
Decision: if either method yields usable validated guides, select 3–4 and export. Only if both come up empty, go to Option 2.
Option 2 — CRISPick precomputed designs
Use when no validated guides exist, you need genome-wide coverage, or you want ranked options.
Step 1 — Resolve the dataset URL
from find_crispick_dataset import find_crispick_dataset
info = find_crispick_dataset("human", cas="SpCas9", application="knockout")
print(info["matches"]) # GRCh38 + GRCh37 dataset URLs
print(info["warning"]) # Cas12a variant warning, if applicable
AsCas12a vs enAsCas12a are different enzymes. Guides for one may not work with the other. The finder matches the exact enzyme token so datasets never cross-contaminate.
Step 2 — Download & extract (files are 50–700 MB; not bundled)
wget '<URL from Step 1>'
gunzip sgRNA_design_*.txt.gz
Step 3 — Filter, rank, select
from select_crispick_sgrnas import select_crispick_sgrnas
picks = select_crispick_sgrnas("sgRNA_design_..._CRISPRko_....txt", "TP53", n=4)
Ranks by Combined Rank (lower = better) by default; rank_by="on_target" or
"off_target" to prioritize efficiency or specificity. Spreads picks across distinct exons for
redundancy. Optional filters: exon=, cut_position_range=, max_target_cut_pct= (knockout).
Column names are resolved defensively (handles both real CRISPick and abbreviated layouts);
see references/crispick_file_format.md. If the gene is absent → Option 3.
Option 3 — De-novo design (last resort)
For genes/organisms not covered above. Follow references/design_rules.md:
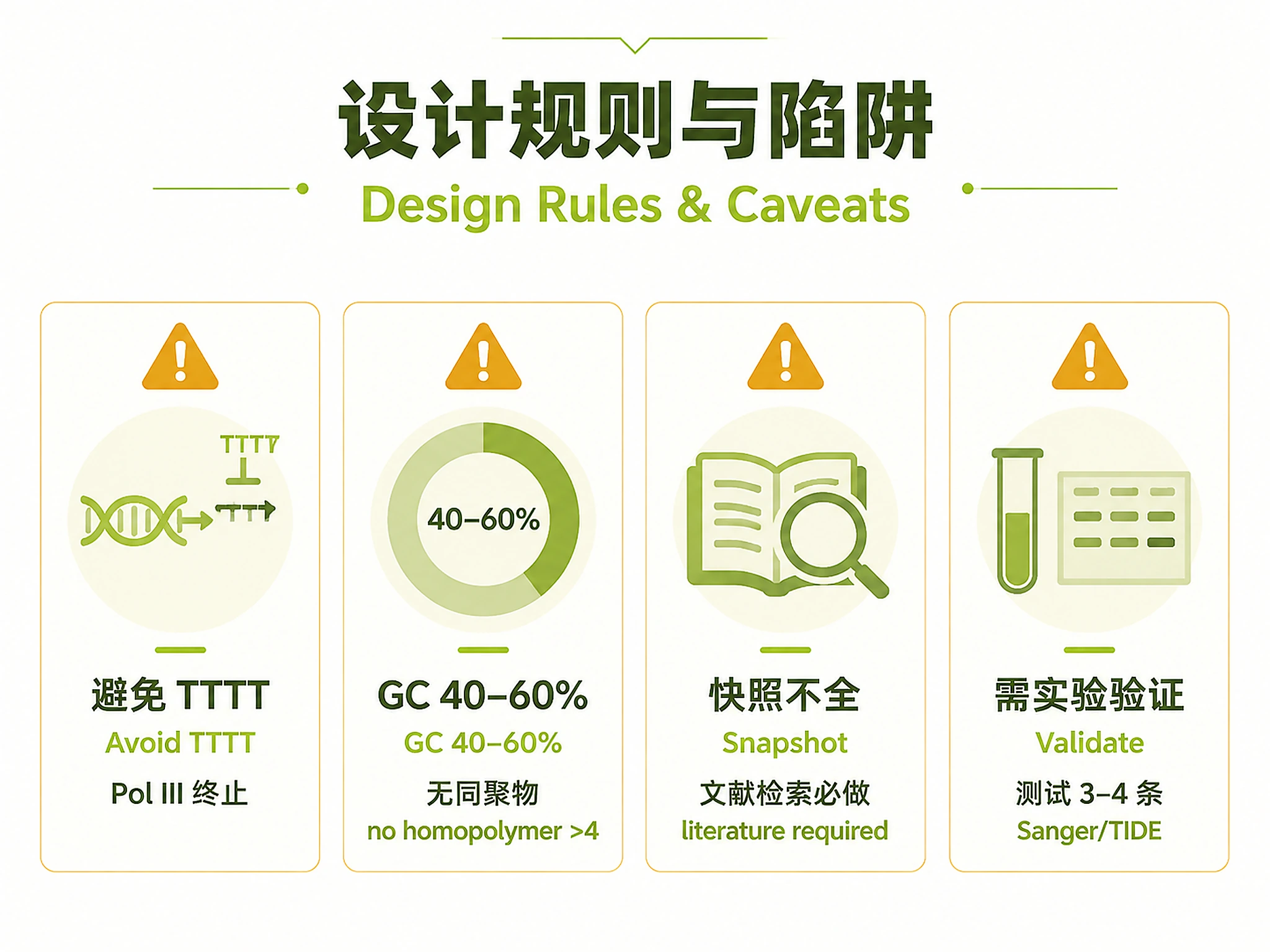
- Length: 20 bp (SpCas9/SaCas9), 23–25 bp (Cas12a). PAM: SpCas9 NGG, SaCas9 NNGRRT, Cas12a TTTV (5').
- GC 40–60%; avoid TTTT and homopolymer runs >4 nt.
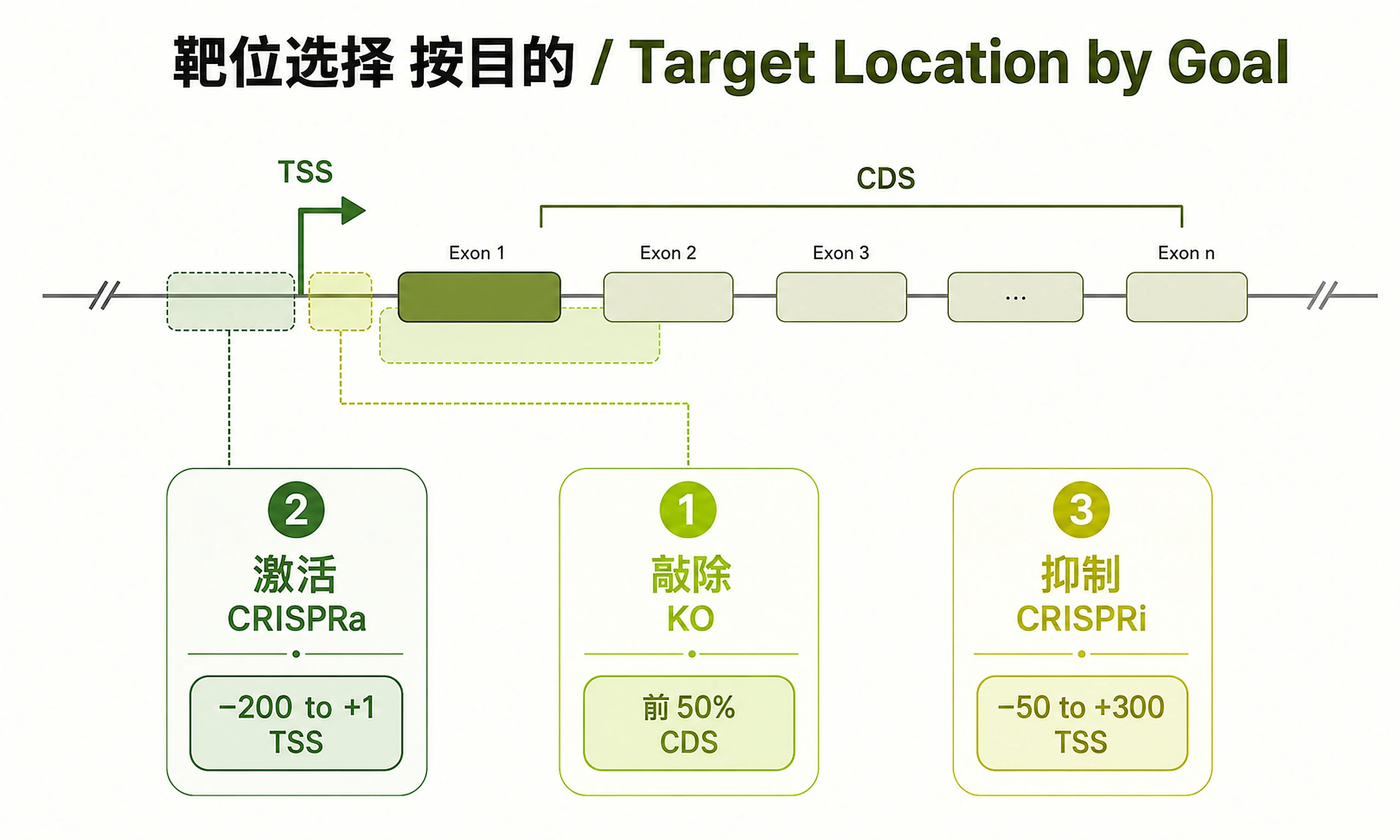
- KO → early exons (first ~50%); CRISPRa → −200 to +1 of TSS; CRISPRi → −50 to +300 of TSS.
from check_design_rules import check_design_rules, format_report
print(format_report(check_design_rules("GAGGTTGTGAGGCGCTGCCC", "SpCas9", pam="AGG")))
This checks rules only — for real off-target assessment use Cas-OFFinder/CRISPOR or CRISPick ranks.
Export (all tiers)
from export_results import from_addgene, from_crispick, export
unified = from_addgene(hits, application="knockout", enzyme="SpCas9") # or from_crispick(...)
export(unified, gene="TP53", tier="Option 1 (validated Addgene)",
outdir="/path/to/results", rationale="Validated guides found via Method 1.")
Universal best practice
Test 3–4 sgRNAs per gene experimentally regardless of predicted scores, and validate edits (Sanger sequencing; TIDE/T7E1 for indels). Prediction scores guide selection but do not replace empirical validation.
Citations & acknowledgments (preserve in user methods)
- Validated guides (Option 1): Addgene (https://www.addgene.org). Cite the PubMed ID of each guide's original publication. Acknowledge: "Validated sgRNA sequences obtained from Addgene."
- CRISPick (Option 2): "Guide designs provided by the CRISPick web tool of the GPP at the Broad Institute."
- Cas9 (SpCas9, SaCas9): Sanson KR, et al. Nat Commun. 2018;9(1):5416. PMID: 30575746.
- Cas12a (AsCas12a, enAsCas12a): DeWeirdt PC, et al. Nat Biotechnol. 2021;39(1):94–104. PMID: 32661438. (Specify which Cas12a variant you used.)
Scientific caveats
- Bundled Addgene/CRISPick files are a fixed snapshot (197 genes / 238 datasets); literature search (Method 2) is mandatory precisely because the snapshot is incomplete.
- Genome build matters: human defaults GRCh38 (GRCh37 also available), mouse GRCm38 — match coordinates to your reference.
- The skill does not perform genome-wide off-target alignment beyond CRISPick's precomputed ranks.
Code preview
scripts/__init__.py
"""sgRNA design skill: helper scripts for the three-tiered workflow."""scripts/check_design_rules.py
"""
Option 3 helper: sanity-check a candidate sgRNA against the de-novo design rules.
This is a lightweight rule checker (length, GC 40-60%, TTTT terminator, >4 homopolymer runs,
and PAM verification for a given enzyme). It is NOT a genome-wide off-target search — use
Cas-OFFinder / CRISPOR or CRISPick's precomputed off-target ranks for real specificity.
"""
from __future__ import annotations
import re
# Enzyme -> (expected protospacer length range, PAM regex, PAM side).
# PAM regex uses IUPAC: N=ACGT, V=ACG, R=AG.
_IUPAC = {"N": "[ACGT]", "V": "[ACG]", "R": "[AG]", "Y": "[CT]", "W": "[AT]",
"S": "[GC]", "K": "[GT]", "M": "[AC]"}
ENZYMES = {
"SpCas9": {"len": (20, 20), "pam": "NGG", "side": "3'"},
"SaCas9": {"len": (20, 21), "pam": "NNGRRT", "side": "3'"},
"AsCas12a": {"len": (23, 25), "pam": "TTTV", "side": "5'"},
"enAsCas12a": {"len": (23, 25), "pam": "TTTV", "side": "5'"},
}
def _pam_to_regex(pam: str) -> str:
return "".join(_IUPAC.get(b, b) for b in pam.upper())
def gc_content(seq: str) -> float:
seq = seq.upper()
if not seq:
return 0.0
return 100.0 * (seq.count("G") + seq.count("C")) / len(seq)
def check_design_rules(protospacer: str, enzyme: str = "SpCas9", pam: str | None = None) -> dict:
"""
Check one candidate protospacer against the de-novo rules.
Parameters
----------
protospacer : str
The guide/protospacer sequence (without the PAM), 5'->3'.
enzyme : str
One of SpCas9, SaCas9, AsCas12a, enAsCas12a.
pam : str, optional
The observed PAM in the genome flanking the protospacer. If given, it is checked
against the enzyme's PAM pattern.
Returns
-------
dict: {'passes': bool, 'checks': {name: (ok, detail)}, 'enzyme': ..., 'gc': float}
"""
seq = protospacer.upper().strip()
spec = ENZYMES.get(enzyme)
checks: dict[str, tuple[bool, str]] = {}
if spec is None:
return {"passes": False, "enzyme": enzyme, "gc": None,
"checks": {"enzyme_known": (False, f"Unknown enzyme '{enzyme}'. "
f"Known: {list(ENZYMES)}")}}
lo, hi = spec["len"]
checks["length"] = (lo <= len(seq) <= hi,
f"{len(seq)} bp (expected {lo}-{hi} for {enzyme})")
gc = gc_content(seq)
checks["gc_content"] = (40.0 <= gc <= 60.0, f"{gc:.0f}% (target 40-60%)")
checks["no_TTTT"] = ("TTTT" not in seq, "TTTT terminator present" if "TTTT" in seq
else "no TTTT run")
homo = re.search(r"(A{5,}|C{5,}|G{5,}|T{5,})", seq)
checks["no_long_homopolymer"] = (homo is None,
f"{homo.group(0)} run" if homo else "no run >4 nt")
checks["valid_bases"] = (re.fullmatch(r"[ACGT]+", seq) is not None,
"non-ACGT characters present" if not re.fullmatch(r"[ACGT]+", seq)
else "all ACGT")scripts/export_results.py
"""
Export selected sgRNAs to a unified CSV + a short markdown summary.
Produces the two deliverables the skill returns for a gene:
<GENE>_selected_sgRNAs.csv - unified table across whichever tier(s) were used
<GENE>_sgRNA_summary.md - which tier was used, why, the picks, and caveats
The unified schema lets validated (Addgene), CRISPick, and de-novo guides sit in one table.
"""
from __future__ import annotations
import os
import pandas as pd
UNIFIED_COLUMNS = [
"gene", "sgRNA_sequence", "source", "application", "enzyme",
"rank_or_score", "exon_or_position", "pam", "citation_or_dataset", "notes",
]
# source values: "validated_addgene" | "crispick" | "de_novo"
def from_addgene(df: pd.DataFrame, application: str = "", enzyme: str = "") -> pd.DataFrame:
"""Map a search_addgene() result into the unified schema."""
rows = []
for _, r in df.iterrows():
cite = f"PMID {r['pubmed_id']}" if r.get("pubmed_id") else ""
if r.get("plasmid_id"):
cite += f"; Addgene #{r['plasmid_id']}"
rows.append({
"gene": r.get("Target Gene", ""),
"sgRNA_sequence": r.get("Target Sequence", ""),
"source": "validated_addgene",
"application": application or r.get("Application", ""),
"enzyme": enzyme or r.get("Cas9 Species", ""),
"rank_or_score": "validated",
"exon_or_position": "",
"pam": "",
"citation_or_dataset": cite,
"notes": f"Depositor: {r.get('Depositor','')}; species: {r.get('Target Species','')}",
})
return pd.DataFrame(rows, columns=UNIFIED_COLUMNS)
def from_crispick(df: pd.DataFrame, gene: str, dataset_url: str = "",
application: str = "", enzyme: str = "") -> pd.DataFrame:
"""Map a select_crispick_sgrnas() result into the unified schema."""
def col(*names):
for n in names:
if n in df.columns:
return n
return None
seq_c = col("sgRNA Sequence", "sgRNA_sequence")
exon_c = col("Exon Number", "Exon_ID")
pos_c = col("sgRNA Cut Position (1-based)", "sgRNA 'Cut' Position")
pam_c = col("PAM Sequence", "PAM")
ds = dataset_url.rsplit("/", 1)[-1] if dataset_url else "CRISPick"
rows = []
for _, r in df.iterrows():
exon_pos = ""
if exon_c and pd.notna(r.get(exon_c)):
exon_pos = f"exon {r[exon_c]}"
if pos_c and pd.notna(r.get(pos_c)):
exon_pos = (exon_pos + f" @ {int(r[pos_c])}").strip()
rows.append({
"gene": gene,
"sgRNA_sequence": r.get(seq_c, "") if seq_c else "",
"source": "crispick",
"application": application,
"enzyme": enzyme,
"rank_or_score": r.get("rank_value", ""),
"exon_or_position": exon_pos,
"pam": r.get(pam_c, "") if pam_c else "",
"citation_or_dataset": ds,
"notes": "CRISPick precomputed; rank lower=better (or score higher=better)",
})
return pd.DataFrame(rows, columns=UNIFIED_COLUMNS)Companion files
| Type | Path | Bytes |
|---|---|---|
| Markdown | references/crispick_file_format.md | 4,037 |
| Markdown | references/design_rules.md | 2,167 |
| Markdown | references/refresh_resources.md | 1,399 |
| CSV | references/resource/addgene_grna_sequences.csv | 54,648 |
| Text | references/resource/CRISPick_download_links.txt | 38,053 |
| Python | scripts/__init__.py | 72 |
| Python | scripts/check_design_rules.py | 3,946 |
| Python | scripts/export_results.py | 6,212 |
| Python | scripts/find_crispick_dataset.py | 6,754 |
| Python | scripts/search_addgene.py | 7,031 |
| Python | scripts/select_crispick_sgrnas.py | 7,165 |
| Markdown | SKILL.md | 7,586 |
| JSON | skill.meta.json | 2,457 |