Omics Dataset Retrieval
Find, catalogue and relevance-audit public omics datasets.
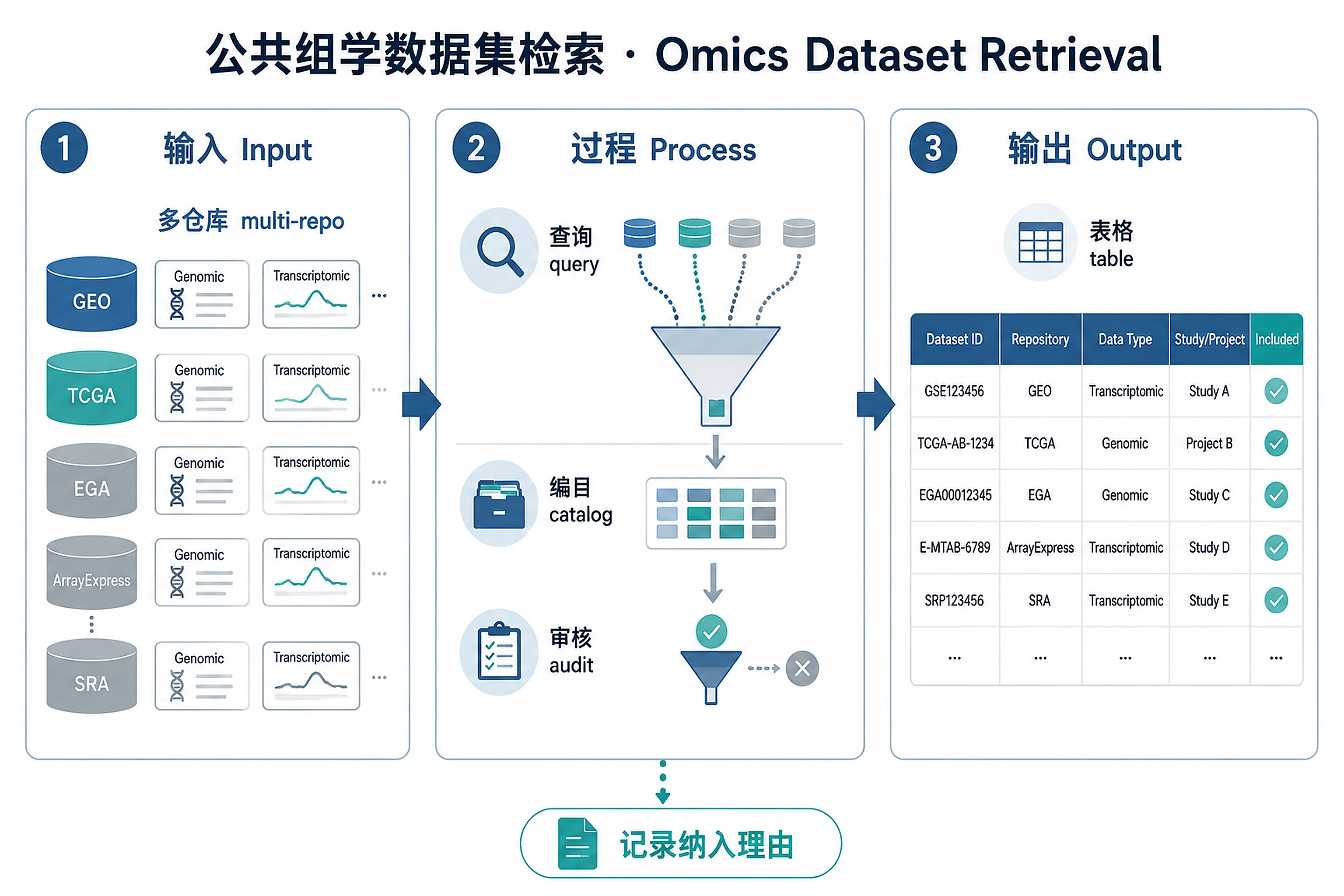
Overview
Problem. Inventory what public data already exists before starting.
Learning goals
- Make surveys traceable — record reasons and evidence
- Inventory vs representative sample — align first
Figures
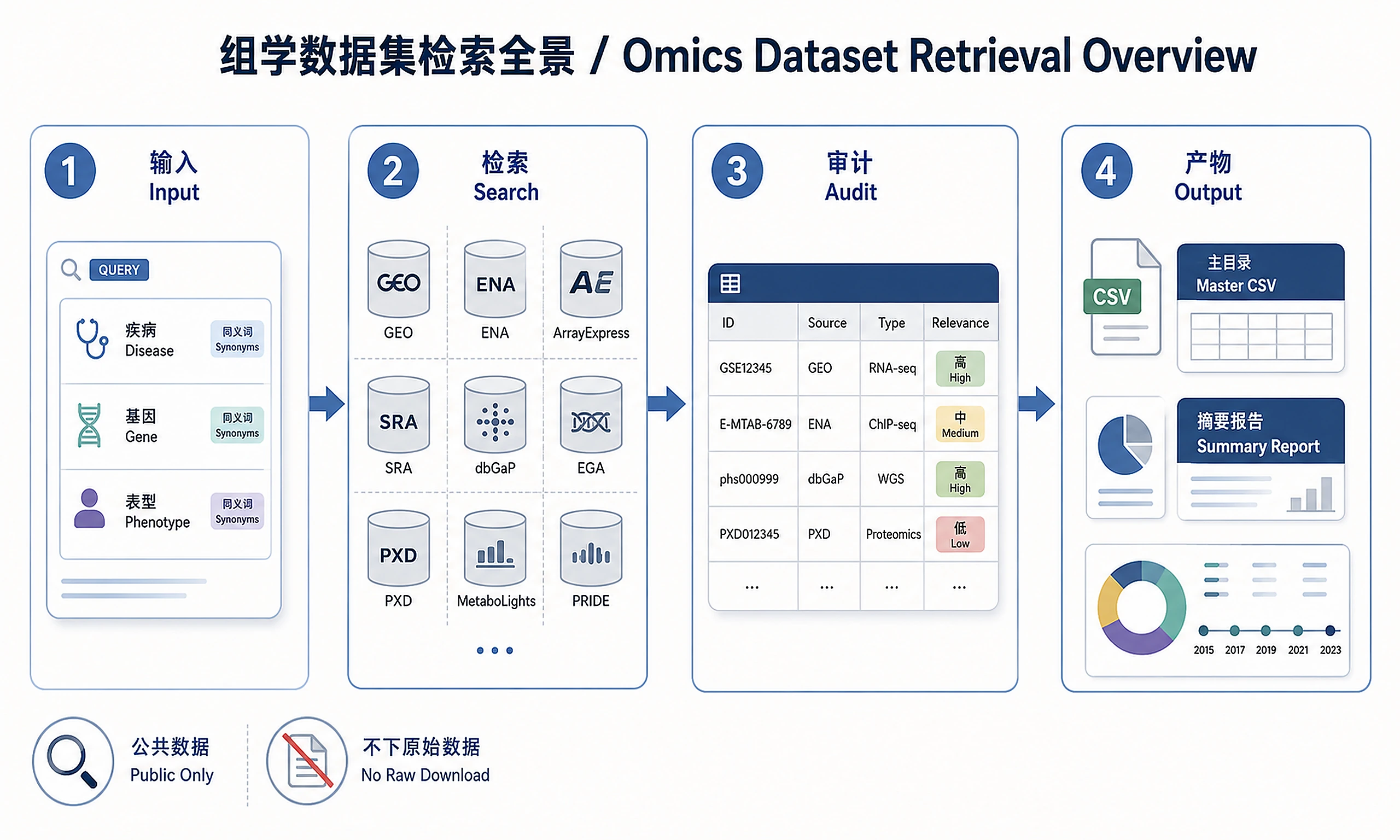
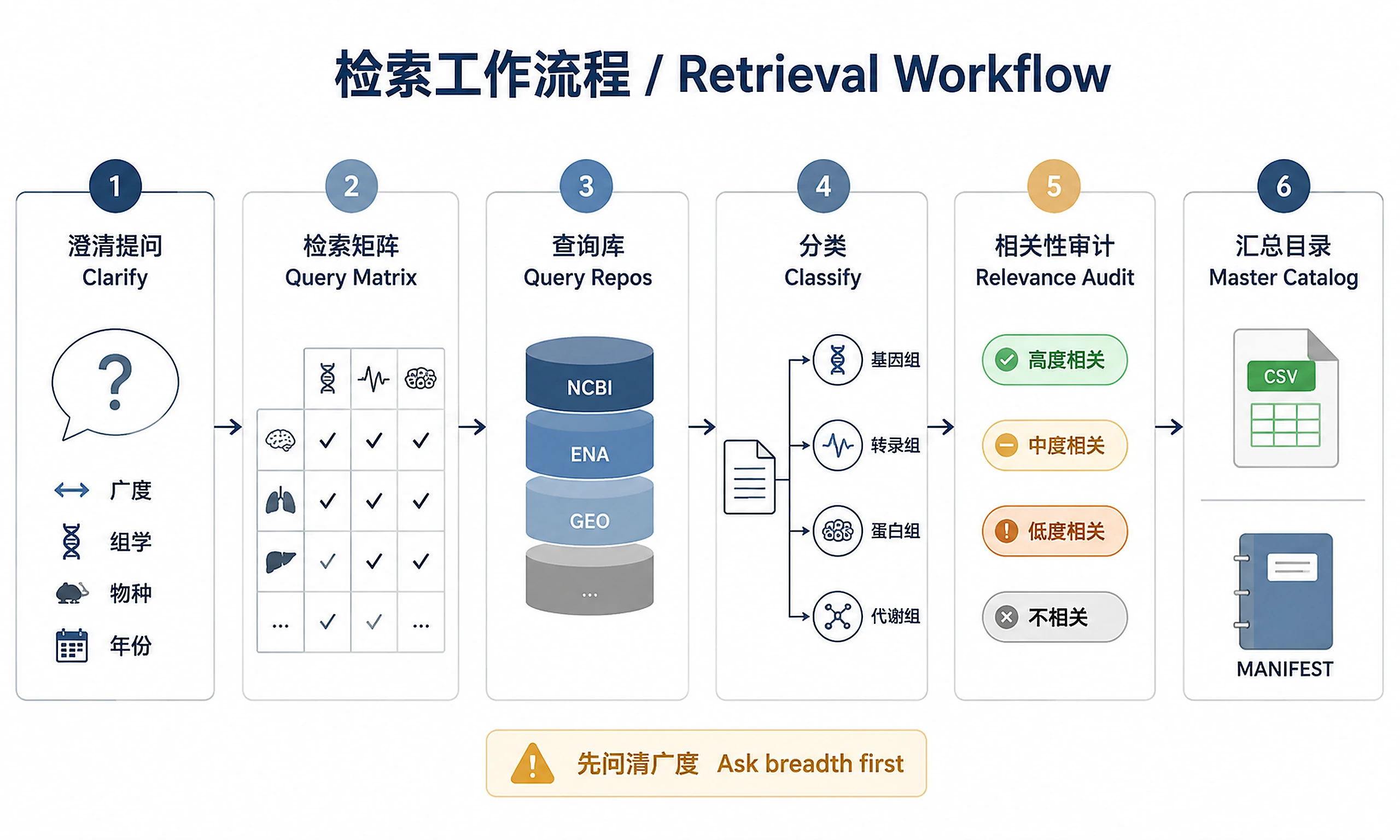


Tutorial
Scope
Systematically retrieve, deduplicate, classify, and relevance-audit publicly available omics datasets for a user-specified disease, phenotype, gene, or biological process. Covers all major omics types (transcriptomics, proteomics, metabolomics, epigenomics, genomics, single-cell, spatial transcriptomics, lipidomics, multi-omics) and the broadest possible set of public repositories. Does not download raw data files or perform downstream analysis.
Inputs
| Parameter | Type | Description |
|---|---|---|
disease_or_topic |
string | Disease name, phenotype, gene, or biological process (e.g., "sickle cell disease", "Alzheimer's disease", "BCL11A") |
synonyms |
list[str] | Alternative names, abbreviations, gene symbols (e.g., ["SCD", "SCA", "HbSS", "sickle cell anemia"]) |
omics_types |
list[str] or "all" | Restrict to specific omics types, or "all" (default) |
organism |
string | "all" (default) — includes human, mouse, and all other organisms; restrict to "human" or "mouse" only if explicitly requested |
year_min |
int | Earliest publication year to include (default: no limit) |
output_dir |
path | Where to save outputs (default: /mnt/results/) |
Outputs
| File | Description |
|---|---|
<disease>_omics_datasets_MASTER.csv |
One comprehensive catalog with all retained candidates and evidence-backed relevance labels |
<disease>_query_manifest.csv |
One row per executed query with repository, exact query, query category, API/source, timestamp, raw hit count, and post-dedup count |
Optional split CSVs, e.g. <disease>_gene_expression_datasets.csv, <disease>_proteomics_datasets.csv |
Only if the user explicitly asks for separate tables; must include the same required relevance audit columns |
<disease>_omics_summary.md |
Markdown summary: search breadth, counts by relevance label / omics type / repository, label definitions, evidence examples, limitations |
<disease>_omics_landscape.png |
Overview figure: donut chart + repository bar + timeline (if requested) |
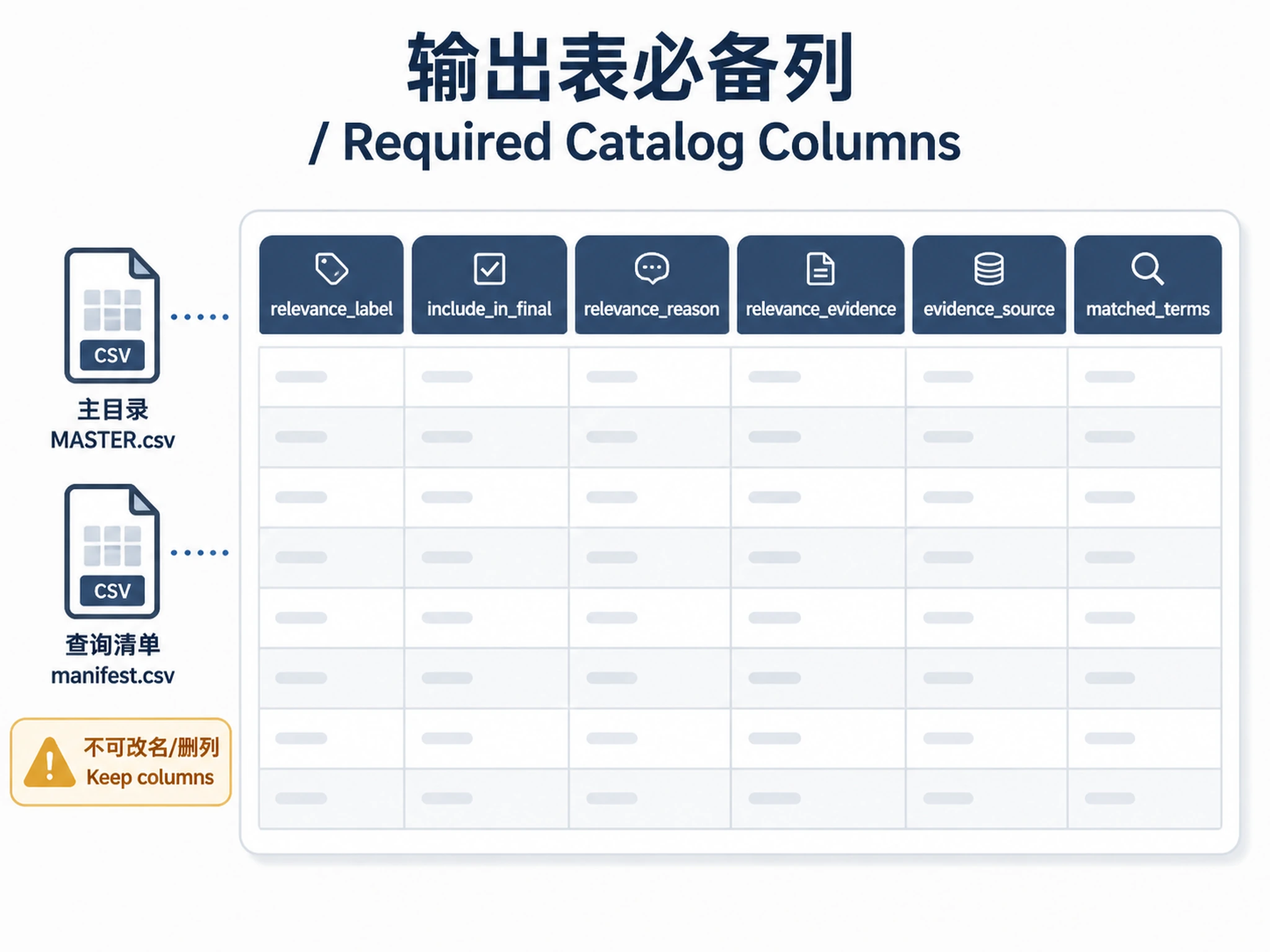
Do not create a default filtered or validated CSV. Save one master CSV by default so that no potentially relevant datasets disappear silently. If the user explicitly asks for a short example list, still use the same columns and state the search breadth in the Markdown summary. If the user asks for separate tables, specific output columns, or a custom table schema, treat those requested columns as additional presentation columns, not an exclusive schema. The required relevance audit columns below must be present in every CSV and Markdown table.
Every CSV and Markdown table must include these relevance audit columns and must not include a numeric relevance score:
| Column | Required content |
|---|---|
relevance_label |
One of direct, adjacent, weak, out_of_scope, false_positive |
include_in_final |
Boolean. true for rows included in final user-facing tables; false for adjacent/weak rows retained only in the master catalog and for excluded candidates |
relevance_reason |
One concise sentence explaining why the label was assigned |
relevance_evidence |
Short title/summary/metadata excerpt supporting the label |
evidence_source |
Field(s) where the evidence came from, e.g. Title, Summary, Publication; never use source_query or Query as relevance evidence |
matched_terms |
Semicolon-separated topic, synonym, tissue, gene, omics, or exclusion terms that matched |
Non-negotiable output rule: never rename, drop, hide, or summarize away these six
columns. Use exactly these column names. Do not rename relevance_label to Relevance.
Do not save a proteomics/gene-expression/custom CSV without these columns. Do not omit them
from a final Markdown table because the user listed other columns; the user's listed columns
are minimum requested fields, not a maximum schema.
The final deliverable may exclude rows where include_in_final == false, but exclusions and
non-final adjacent/weak rows must be auditable. Keep them in the master audit catalog or an
excluded-candidates CSV, and summarize counts and examples in the Markdown report. By default,
primary final split tables include direct rows only; include adjacent or weak rows only
when the user explicitly asks for a broader final table.
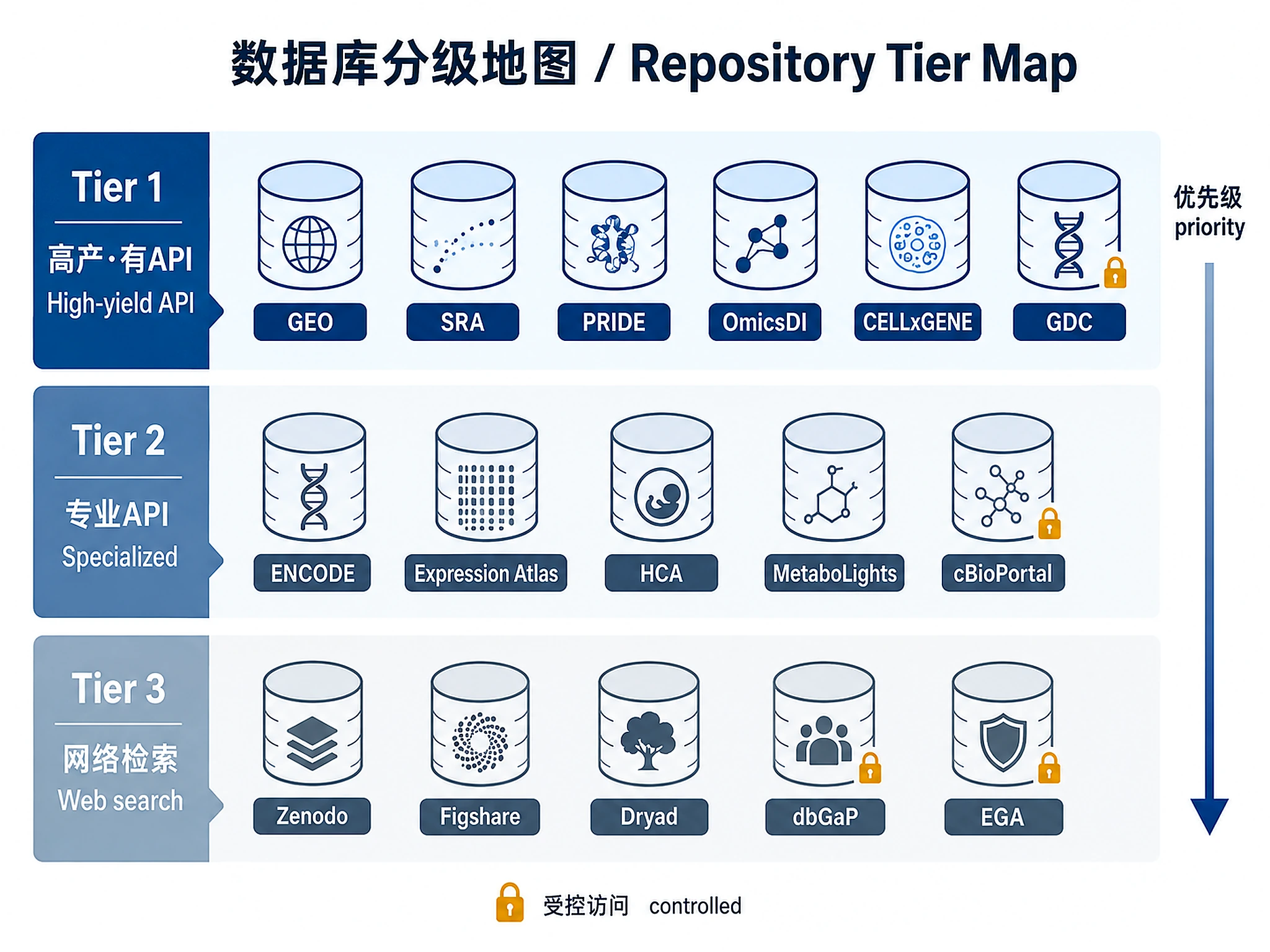
Repository Coverage Map
Work through repositories in priority order. Tier 1 repositories have programmatic APIs and are the highest-yield sources. Tier 2 have APIs but are more specialized. Tier 3 require web search or manual curation. In comprehensive inventory mode, attempt all tiers because rare diseases often have data only in Tier 2/3 repositories. In representative examples mode, start with Tier 1 and add Tier 2/3 only as needed to cover the requested omics types.
Tier 1 — High-yield, programmatic APIs (query first)
| Repository | Omics Focus | API Base URL | Notes |
|---|---|---|---|
| GEO (NCBI) | All transcriptomics, epigenomics | https://eutils.ncbi.nlm.nih.gov/entrez/eutils/ |
Largest source; run 20–40 targeted queries |
| SRA (NCBI) | Raw sequencing reads | https://eutils.ncbi.nlm.nih.gov/entrez/eutils/ (db=sra) |
Complements GEO; finds studies not deposited as GEO series |
| ArrayExpress / BioStudies (EBI) | Transcriptomics, functional genomics | https://www.ebi.ac.uk/biostudies/api/v1/search |
European mirror of GEO; many unique studies |
| PRIDE / ProteomeXchange | Proteomics (MS) | https://www.ebi.ac.uk/pride/ws/archive/v2/projects/search |
Primary proteomics repository |
| OmicsDI (aggregator) | Multi-omics aggregator | https://www.omicsdi.org/ws/dataset/search |
Covers MetaboLights, MassIVE, GNPS, Metabolomics Workbench, ArrayExpress, GEO, PRIDE — use to catch metabolomics and avoid re-querying individual repos |
| CZ CELLxGENE | scRNA-seq, spatial | https://api.cellxgene.cziscience.com/curation/v1/collections |
33M+ cells; query by disease using cellxgene_census Python package |
| GDC / TCGA (NCI) | Cancer multi-omics | https://api.gdc.cancer.gov/ |
Best for cancer diseases; covers TCGA, TARGET, CGCI, CPTAC-GDC |
Tier 2 — Specialized APIs (query when relevant to disease/omics type)
| Repository | Omics Focus | API / Access | When to use |
|---|---|---|---|
| ENCODE | Epigenomics (ChIP-seq, ATAC-seq, RNA-seq) | https://www.encodeproject.org/search/?format=json |
Regulatory genomics; TF binding; chromatin accessibility |
| Expression Atlas (EBI) | Bulk + single-cell RNA-seq | https://www.ebi.ac.uk/gxa/json/experiments |
Curated, baseline + differential expression experiments |
| Human Cell Atlas (HCA) | scRNA-seq, spatial | https://service.azul.data.humancellatlas.org/index/projects |
Reference cell type atlases; healthy tissue baselines |
| Metabolomics Workbench (NIH) | Metabolomics, lipidomics | https://www.metabolomicsworkbench.org/rest/study/study_id/ST/named_json |
NIH-funded metabolomics; REST API available |
| MetaboLights (EBI) | Metabolomics | https://www.ebi.ac.uk/metabolights/ws/studies/ |
European metabolomics repository |
| MassIVE / GNPS | Metabolomics, lipidomics | https://massive.ucsd.edu/ProteoSAFe/datasets.jsp |
MS-based metabolomics; use OmicsDI to search |
| jPOST | Proteomics (MS) | https://repository.jpostdb.org/search |
Japanese proteomics repository; ProteomeXchange member |
| iProX | Proteomics (MS) | https://www.iprox.cn/page/project.html |
Chinese proteomics repository; ProteomeXchange member |
| cBioPortal | Cancer multi-omics | https://www.cbioportal.org/api/ |
Cancer genomics; mutation, CNA, expression, methylation |
| EpiRR / IHEC | Epigenomics reference | https://www.ebi.ac.uk/epirr/api/ |
IHEC reference epigenomes; healthy tissue baselines |
| Human Protein Atlas (HPA) | Proteomics, RNA-seq | https://www.proteinatlas.org/api/ |
Tissue/cell-type protein and RNA expression |
| ENA (EBI) | Raw sequencing | https://www.ebi.ac.uk/ena/portal/api/search |
European mirror of SRA; raw reads |
Tier 3 — Web search + manual curation (always attempt in comprehensive mode)
| Repository | Omics Focus | Search Strategy |
|---|---|---|
| Zenodo | All types | WebSearch: "{disease}" omics dataset site:zenodo.org |
| Figshare | All types | WebSearch: "{disease}" RNA-seq proteomics site:figshare.com |
| Dryad | All types | WebSearch: "{disease}" omics data site:datadryad.org |
| OSF (Open Science Framework) | All types | WebSearch: "{disease}" omics dataset site:osf.io |
| Harvard Dataverse | All types | WebSearch: "{disease}" omics dataset site:dataverse.harvard.edu |
| Synapse (Sage Bionetworks) | Neuroscience, cancer | WebSearch: "{disease}" omics dataset site:synapse.org |
| ICGC Data Portal | Cancer genomics | WebSearch: "{disease}" ICGC site:dcc.icgc.org |
| CPTAC (NCI) | Cancer proteomics | WebSearch: "{disease}" CPTAC proteomics site:proteomics.cancer.gov |
| AWS Open Data Registry | All types | WebSearch: "{disease}" omics site:registry.opendata.aws |
| dbGaP (NIH, controlled) | Genomics, WGS | WebSearch: "{disease}" WGS genomics site:ncbi.nlm.nih.gov/gap |
| EGA (EBI, controlled) | Genomics, WGS | WebSearch: "{disease}" genome sequencing site:ega-archive.org |
| JGA (Japan, controlled) | Genomics | WebSearch: "{disease}" genomics site:ddbj.nig.ac.jp/jga |
| UK Biobank | Population genomics | WebSearch: "{disease}" UK Biobank omics |
| FinnGen | Population genomics | WebSearch: "{disease}" FinnGen GWAS |
Workflow
Step 1 — Ask upfront clarification questions (MANDATORY before any search)
This step is not optional. Before running any queries, use AskUserQuestion to collect
the information below. Do not assume defaults — the answers materially affect which repositories
are queried, how many results are returned, and how the relevance audit is tuned.
Ask ALL of the following in a single AskUserQuestion call (skip any already answered in the
user's initial message, but always ask the rest):
Q1 — Search breadth
- Should the result be a comprehensive inventory or only a few representative examples?
- Comprehensive inventory (default) — search as broadly as possible and keep all plausible candidates in the master CSV with relevance labels.
- Representative examples — return only a small curated sample, still with relevance labels and evidence. Ask the user for an approximate count if they do not provide one.
Q2 — Disease / topic (if not already provided)
- What disease, phenotype, gene, or biological process should we search for?
- Any synonyms, abbreviations, or alternative names to include? (e.g., "SCD", "SCA", "HbSS" for sickle cell disease — more synonyms = better recall)
- Any key genes or pathways central to this topic? (used to build additional targeted GEO queries beyond the disease name alone)
Q3 — Omics types
- Which omics types should be included?
- All types (default) — transcriptomics, proteomics, metabolomics, epigenomics, genomics, single-cell, spatial, multi-omics
- Transcriptomics only — bulk RNA-seq, microarray, scRNA-seq, spatial
- Epigenomics only — ChIP-seq, ATAC-seq, methylation, CUT&RUN, Hi-C
- Proteomics / metabolomics only — MS proteomics, metabolomics, lipidomics
- Custom — user specifies which types to include
Q4 — Organism
- Which organisms should be included?
- All organisms (default) — human, mouse, and any other species
- Human only — Homo sapiens studies only
- Specific species — user specifies (e.g., mouse only, zebrafish only)
Q5 — Year range
- Any restriction on publication/deposit year?
- No restriction (default) — all years
- Recent only — e.g., 2018 onwards, 2020 onwards
- Custom range — user specifies start and/or end year
Q6 — Controlled-access repositories
- Should we include controlled-access repositories (dbGaP, EGA, JGA)? These require institutional data access agreements to download data, but we can still catalog their existence and metadata.
- Yes, include and flag them (default) — catalog with "Controlled" label
- No, open-access only — skip dbGaP, EGA, JGA entirely
Q7 — Goal / output format
- What is the primary goal?
- Browse and select (default) — CSV catalog + Markdown summary for review
- Landscape overview — also generate a visual overview figure (donut + timeline + repository bar)
- Download and analyze — also include direct download links and file formats where available
- All of the above
Q8 — Tissue or cell type focus (optional but significantly improves recall)
- Are there specific tissues, cell types, or sample sources to prioritize? (e.g., "whole blood", "brain cortex", "CD34+ HSCs", "plasma", "tumor") These are added as targeted search terms to GEO and other repositories. If none specified, broad disease-name queries are used.
Q9 — Final table inclusion
- Which relevance labels should appear in final user-facing tables?
- Direct only (default) — final split tables include only datasets with direct disease/topic evidence; adjacent and weak candidates stay in the master audit catalog.
- Direct + adjacent — include direct rows plus mechanistically related adjacent rows in final tables.
- Direct + adjacent + weak — include broad or tangential weak rows too; use only when the user wants maximum recall in the final tables.
Q10 — Raw-read granularity
- How should SRA/ENA raw-read repositories be represented?
- Study/project level (default) — aggregate raw reads by study/project accession to avoid duplicate SRR/SRX run inflation.
- Run level — expand individual raw runs only if the user explicitly wants run-level accessions.
Only proceed to Step 2 once all answers are collected. If the user provides partial information upfront (e.g., just the disease name), ask only the remaining unanswered questions.
Step 2 — Build search term matrix
Build the search term matrix according to the user's requested breadth.
Do not use a disease-specific hardcoded query manifest. Use a fixed query-generation protocol so the behavior is consistent while still adapting to the user's disease/topic.
For comprehensive inventory mode, generate a broad set of search queries by combining:
- Primary disease name + synonyms
- Omics-type-specific terms (RNA-seq, microarray, ChIP-seq, ATAC-seq, scRNA-seq, proteomics, metabolomics, methylation, WGS, SNP array, CUT&RUN, Hi-C, spatial transcriptomics, lipidomics)
- Tissue/cell-type terms relevant to the disease
- Clinical context terms (treatment, crisis, biomarker, pediatric, longitudinal)
- Key gene/pathway terms central to the disease
For representative examples mode, use the same term categories but run fewer high-precision queries, prefer records with strong metadata and publication links, and do not describe the result as exhaustive.
For every query, record provenance fields on each returned row:
| Field | Meaning |
|---|---|
source_repository |
Repository or aggregator queried |
source_query |
Exact query string or structured API filters used |
source_query_category |
One of disease_exact, synonym, omics_type, tissue_cell_type, clinical_context, gene_pathway, controlled_access, manual_web |
source_api |
API/tool endpoint or web search source |
retrieved_at |
Timestamp for the retrieval run |
n_raw_hits |
Number of raw hits returned by that query before cross-query deduplication |
n_records_after_dedup |
Number of unique records from that query category/source retained after accession-level deduplication |
Save these provenance fields as <disease>_query_manifest.csv. source_query and related
query fields explain why a candidate was retrieved, but they are provenance only. Never use
source_query, source_query_category, source_repository, source_api, retrieved_at, or
Query as positive relevance evidence for direct or adjacent labels.
query_manifest_rows = []
def add_query_manifest_row(source_repository, source_query, source_query_category,
source_api, retrieved_at, n_raw_hits, n_records_after_dedup):
query_manifest_rows.append({
"source_repository": source_repository,
"source_query": source_query,
"source_query_category": source_query_category,
"source_api": source_api,
"retrieved_at": retrieved_at,
"n_raw_hits": n_raw_hits,
"n_records_after_dedup": n_records_after_dedup,
})
GEO query syntax tips:
- Use
[Title]field tag for high-precision hits:"sickle cell"[Title] AND "RNA-seq" - Use
[DataSet Type]for omics filtering:"expression profiling by array"[DataSet Type] - Use
retmax=100per query; run 20–40 queries to maximize recall - Deduplicate by GEO UID across all queries
Step 3 — Query GEO (NCBI E-utilities) — PRIMARY SOURCE
import requests, time, pandas as pd
BASE = "https://eutils.ncbi.nlm.nih.gov/entrez/eutils/"
# Comprehensive mode: build 20-40 queries combining disease + omics + tissue + gene terms.
# Representative mode: run a smaller high-precision subset and report that it is not exhaustive.
# Example for any disease:
search_queries = {
"title_rnaseq": f'"{disease}"[Title] AND "RNA-seq"',
"title_scrna": f'"{disease}"[Title] AND "single cell"',
"title_array": f'"{disease}"[Title] AND "expression profiling by array"[DataSet Type]',
"title_chip": f'"{disease}" AND "ChIP-seq"',
"title_atac": f'"{disease}" AND "ATAC-seq"',
"title_methyl": f'"{disease}" AND "methylation"',
"title_wgs": f'"{disease}" AND "whole genome sequencing"',
"title_proteom": f'"{disease}" AND "proteomics"',
"title_metabolom": f'"{disease}" AND "metabolomics"',
# Add synonym-based queries:
# f'"{synonym}"[Title] AND "RNA-seq"' for each synonym
# Add tissue-specific queries:
# f'"{disease}" AND "{tissue_term}"' for key tissues
# Add gene-specific queries:
# f'"{disease}" AND "{key_gene}"' for key disease genes
}
all_ids = set()
for label, query in search_queries.items():
r = requests.get(f"{BASE}esearch.fcgi",
params={"db": "gds", "term": query, "retmax": 100, "retmode": "json"})
ids = r.json().get("esearchresult", {}).get("idlist", [])
all_ids.update(ids)
time.sleep(0.34) # Stay under NCBI rate limit (3 req/sec without API key)
# Fetch summaries in batches of 20
records = []
for i in range(0, len(list(all_ids)), 20):
batch = list(all_ids)[i:i+20]
r = requests.get(f"{BASE}esummary.fcgi",
params={"db": "gds", "id": ",".join(batch), "retmode": "json"})
result = r.json().get("result", {})
for uid in result.get("uids", []):
item = result[uid]
records.append({
"Accession": item.get("accession", ""),
"Title": item.get("title", ""),
"GEO_Type": item.get("gdstype", ""),
"Organism": item.get("taxon", ""),
"N_Samples": item.get("n_samples", ""),
"Date": item.get("pdat", ""),
"Summary": item.get("summary", "")[:500],
"Repository": "GEO",
})
time.sleep(0.34)
df_geo = pd.DataFrame(records)
# Filter: GSE/GDS accessions only (exclude GPL platform records)
# By default, keep ALL organisms. Only filter by organism if user explicitly requested a specific species.
# Example organism filter (apply only if requested):
# df_geo = df_geo[df_geo["Organism"].str.contains("Homo sapiens", na=False)]
df_geo = df_geo[df_geo["Accession"].str.startswith(("GSE", "GDS"))]
NCBI API key: Set api_key param to increase rate limit to 10 req/sec (register free at NCBI).
Step 4 — Query SRA (NCBI) for raw sequencing studies not in GEO
# SRA catches studies deposited as raw reads without a GEO series.
# Default: aggregate at study/project level to avoid SRR/SRX run-level inflation.
# Expand to individual runs only if the user explicitly requested run-level raw reads.
RAW_READ_GRANULARITY = "study_project" # or "run_level" if explicitly requested
EXPAND_RAW_RUNS = RAW_READ_GRANULARITY == "run_level"
sra_records = []
for query in [disease] + synonyms[:3]:
# Default: search all organisms. Add AND "Homo sapiens"[Organism] only if user requested human-only.
r = requests.get(f"{BASE}esearch.fcgi",
params={"db": "sra", "term": f'"{query}"',
"retmax": 100, "retmode": "json"})
ids = r.json().get("esearchresult", {}).get("idlist", [])
if ids:
r2 = requests.get(f"{BASE}esummary.fcgi",
params={"db": "sra", "id": ",".join(ids), "retmode": "json"})
result = r2.json().get("result", {})
for uid in result.get("uids", []):
item = result[uid]
exp_acc = item.get("experiment", {}).get("acc", "")
study_acc = (
item.get("study", {}).get("acc", "")
or item.get("bioproject", "")
or exp_acc
)
runs_obj = item.get("runs", {})
runs = runs_obj.get("Run", []) if isinstance(runs_obj, dict) else []
if isinstance(runs, dict):
runs = [runs]
run_accs = [run.get("acc", "") for run in runs if run.get("acc")]
accession = ";".join(run_accs) if EXPAND_RAW_RUNS and run_accs else study_acc
sra_records.append({
"Accession": accession,
"Raw_Run_Accessions": ";".join(run_accs),
"Raw_Read_Granularity": RAW_READ_GRANULARITY,
"Title": item.get("title", ""),
"GEO_Type": item.get("exptype", ""),
"Organism": item.get("organism", {}).get("ScientificName", "unknown"),
"N_Samples": len(run_accs) if run_accs else (runs_obj.get("@total", "") if isinstance(runs_obj, dict) else ""),
"Date": item.get("createdate", ""),
"Summary": item.get("summary", "")[:500],
"Repository": "SRA",
})
time.sleep(0.34)
Step 5 — Query ArrayExpress / BioStudies (EBI)
# BioStudies API — covers ArrayExpress functional genomics collection
# Source: https://www.ebi.ac.uk/biostudies/api/v1/search
biostudies_url = "https://www.ebi.ac.uk/biostudies/api/v1/search"
biostudies_records = []
for keyword in [disease] + synonyms[:2]:
r = requests.get(biostudies_url,
params={"query": keyword, "collection": "arrayexpress",
"pageSize": 100, "page": 1},
headers={"Accept": "application/json"}, timeout=60)
if r.status_code == 200:
data = r.json()
for hit in data.get("hits", []):
biostudies_records.append({
"Accession": hit.get("accession", ""),
"Title": hit.get("title", ""),
"GEO_Type": hit.get("type", ""),
"Organism": "; ".join([o.get("scientificName","") for o in hit.get("organism",[])]),
"N_Samples": hit.get("numberOfSamples", ""),
"Date": hit.get("releaseDate", ""),
"Summary": hit.get("description", "")[:500],
"Repository": "ArrayExpress/BioStudies",
})
time.sleep(1)
df_biostudies = pd.DataFrame(biostudies_records).drop_duplicates(subset="Accession")
# Exclude GEO mirror accessions (E-GEOD- prefix = same study already captured via GEO)
# Keep all organisms by default. Apply organism filter only if user explicitly requested a specific species.
# Example: df_biostudies = df_biostudies[df_biostudies["Organism"].str.contains("Homo sapiens|human", case=False, na=False)]
df_biostudies = df_biostudies[~df_biostudies["Accession"].str.startswith("E-GEOD")]
Step 6 — Query PRIDE / ProteomeXchange (proteomics)
# PRIDE REST API v2
# Source: https://www.ebi.ac.uk/pride/ws/archive/v2/projects/search
pride_url = "https://www.ebi.ac.uk/pride/ws/archive/v2/projects/search"
pride_records = []
for keyword in [disease] + synonyms:
r = requests.get(pride_url,
params={"keyword": keyword, "pageSize": 100, "page": 0},
headers={"Accept": "application/json"}, timeout=60)
if r.status_code == 200:
data = r.json()
# Handle both list and dict response formats
projects = data if isinstance(data, list) else data.get("_embedded", {}).get("compactprojects", [])
for p in projects:
pride_records.append({
"Accession": p.get("accession", ""),
"Title": p.get("title", ""),
"GEO_Type": "Proteomics (MS)",
"Organism": "; ".join(p.get("organisms", [])),
"N_Samples": p.get("numberOfSamples", ""),
"Date": p.get("submissionDate", ""),
"Summary": p.get("projectDescription", "")[:500],
"Repository": "PRIDE",
})
time.sleep(1)
df_pride = pd.DataFrame(pride_records).drop_duplicates(subset="Accession")
# Keep all organisms by default. Apply organism filter only if user explicitly requested a specific species.
# Example: df_pride = df_pride[df_pride["Organism"].str.contains("Homo sapiens|human", case=False, na=False)]
Also check jPOST and iProX (ProteomeXchange members not always in PRIDE search):
# jPOST REST API
# Source: https://repository.jpostdb.org/search
jpost_records = []
for keyword in [disease] + synonyms[:2]:
r = requests.get("https://repository.jpostdb.org/search",
params={"keyword": keyword, "format": "json"}, timeout=30)
if r.status_code == 200:
for p in r.json().get("projects", []):
jpost_records.append({
"Accession": p.get("projectId", ""),
"Title": p.get("title", ""),
"GEO_Type": "Proteomics (MS)",
"Organism": p.get("organism", ""),
"N_Samples": "",
"Date": p.get("releaseDate", ""),
"Summary": p.get("description", "")[:500],
"Repository": "jPOST",
})
time.sleep(1)
Step 7 — Query OmicsDI (metabolomics aggregator)
OmicsDI is the single best entry point for metabolomics — it aggregates MetaboLights, MassIVE, GNPS, Metabolomics Workbench, and Metabolon. Filter by source to avoid GEO duplicates.
# OmicsDI aggregator
# Source: https://www.omicsdi.org/ws/dataset/search
omicsdi_url = "https://www.omicsdi.org/ws/dataset/search"
omicsdi_records = []
METABOLOMICS_REPOS = {"MetaboLights", "MassIVE", "GNPS", "Metabolon",
"Metabolomics Workbench", "HMDB", "Lipidmaps"}
for keyword in [disease] + synonyms[:2]:
r = requests.get(omicsdi_url,
params={"query": keyword, "size": 100, "start": 0},
headers={"Accept": "application/json"}, timeout=60)
if r.status_code == 200:
for ds in r.json().get("datasets", []):
repo = ds.get("source", "")
if repo in METABOLOMICS_REPOS:
omicsdi_records.append({
"Accession": ds.get("id", ""),
"Title": ds.get("name", ""),
"GEO_Type": "Metabolomics",
"Organism": "; ".join(ds.get("organisms", {}).get("name", [])),
"N_Samples": "",
"Date": ds.get("publicationDate", ""),
"Summary": ds.get("description", "")[:500],
"Repository": repo,
})
time.sleep(1)
df_metabolomics = pd.DataFrame(omicsdi_records).drop_duplicates(subset="Accession")
Also query Metabolomics Workbench REST API directly for NIH-funded studies:
# Metabolomics Workbench REST API
# Source: https://www.metabolomicsworkbench.org/tools/mw_rest.php
mw_url = "https://www.metabolomicsworkbench.org/rest/study/study_title"
for keyword in [disease] + synonyms[:2]:
r = requests.get(f"{mw_url}/{requests.utils.quote(keyword)}/summary/json", timeout=30)
if r.status_code == 200 and r.text.strip():
# Response is a dict of study_id -> study metadata
for sid, meta in r.json().items():
omicsdi_records.append({
"Accession": sid,
"Title": meta.get("study_title", ""),
"GEO_Type": "Metabolomics",
"Organism": meta.get("subject_species", ""),
"N_Samples": meta.get("subject_count", ""),
"Date": meta.get("submit_date", ""),
"Summary": meta.get("study_summary", "")[:500],
"Repository": "Metabolomics Workbench",
})
Step 8 — Query CZ CELLxGENE (single-cell)
# CZ CELLxGENE Collections API
# Source: https://api.cellxgene.cziscience.com/curation/v1/collections
cxg_url = "https://api.cellxgene.cziscience.com/curation/v1/collections"
r = requests.get(cxg_url, headers={"Accept": "application/json"}, timeout=60)
cxg_records = []
if r.status_code == 200:
for col in r.json():
title = col.get("name", "")
desc = col.get("description", "")
# Filter by disease keywords
text = (title + " " + desc).lower()
if any(kw.lower() in text for kw in [disease] + synonyms):
cxg_records.append({
"Accession": col.get("collection_id", ""),
"Title": title,
"GEO_Type": "scRNA-seq",
"Organism": "; ".join(sorted({d.get("organism","") for d in col.get("datasets",[]) if d.get("organism")})) or "unknown",
"N_Samples": col.get("cell_count", ""),
"Date": col.get("published_at", ""),
"Summary": desc[:500],
"Repository": "CZ CELLxGENE",
})
# Also use cellxgene_census Python package for cell-level metadata queries:
# import cellxgene_census
# census = cellxgene_census.open_soma()
# obs = census["census_data"]["homo_sapiens"].obs.read(
# value_filter=f'disease == "{disease_ontology_term}"'
# ).concat().to_pandas()
Step 9 — Query GDC / TCGA (cancer diseases)
Only relevant for cancer diseases. Skip for non-cancer diseases.
# GDC REST API
# Source: https://api.gdc.cancer.gov/
gdc_url = "https://api.gdc.cancer.gov/projects"
r = requests.get(gdc_url,
params={"filters": json.dumps({"op": "in", "content": {
"field": "disease_type", "value": [disease] + synonyms}}),
"fields": "project_id,name,disease_type,primary_site,summary",
"format": "json", "size": 100},
timeout=60)
# Parse and add to records with Repository = "GDC/TCGA"
Step 10 — Query ENCODE (epigenomics)
Only relevant when the disease has known epigenomic/regulatory components.
# ENCODE REST API — rate limit: 10 req/sec
# Source: https://www.encodeproject.org/help/rest-api
encode_url = "https://www.encodeproject.org/search/"
r = requests.get(encode_url,
params={"searchTerm": disease, "type": "Experiment",
"status": "released", "format": "json", "limit": 100},
headers={"Accept": "application/json"}, timeout=60)
if r.status_code == 200:
for exp in r.json().get("@graph", []):
encode_records.append({
"Accession": exp.get("accession", ""),
"Title": exp.get("description", ""),
"GEO_Type": exp.get("assay_title", ""),
"Organism": exp.get("organism", {}).get("scientific_name", ""),
"N_Samples": len(exp.get("replicates", [])),
"Date": exp.get("date_released", ""),
"Summary": exp.get("biosample_summary", "")[:500],
"Repository": "ENCODE",
})
Step 11 — Query Expression Atlas (EBI)
# Expression Atlas REST API
# Source: https://www.ebi.ac.uk/gxa/json/experiments
# Default: no species filter — returns all organisms. Add species param only if user requested a specific organism.
atlas_url = "https://www.ebi.ac.uk/gxa/json/experiments"
r = requests.get(atlas_url, timeout=60) # omit species= to get all organisms
atlas_records = []
if r.status_code == 200:
for exp in r.json().get("experiments", []):
text = (exp.get("experimentDescription","") + " " +
" ".join(exp.get("factors",[])) + " " +
" ".join(exp.get("experimentalFactors",[]))).lower()
if any(kw.lower() in text for kw in [disease] + synonyms):
atlas_records.append({
"Accession": exp.get("experimentAccession", ""),
"Title": exp.get("experimentDescription", ""),
"GEO_Type": exp.get("experimentType", ""),
"Organism": "; ".join(exp.get("species", [])) or "unknown",
"N_Samples": exp.get("numberOfAssays", ""),
"Date": exp.get("lastUpdate", ""),
"Summary": "",
"Repository": "Expression Atlas",
})
Step 12 — Web search for Tier 3 repositories
Use the WebSearch tool for repositories without disease-specific APIs. Run all of these:
# Open repositories
WebSearch: "{disease}" omics dataset site:zenodo.org
WebSearch: "{disease}" RNA-seq proteomics site:figshare.com
WebSearch: "{disease}" omics data site:datadryad.org
WebSearch: "{disease}" omics dataset site:osf.io
WebSearch: "{disease}" omics dataset site:dataverse.harvard.edu
WebSearch: "{disease}" omics dataset site:synapse.org
# Controlled-access repositories
WebSearch: "{disease}" WGS genomics site:ncbi.nlm.nih.gov/gap
WebSearch: "{disease}" genome sequencing site:ega-archive.org
WebSearch: "{disease}" genomics site:ddbj.nig.ac.jp/jga
# Consortium/specialized
WebSearch: "{disease}" ICGC genomics dataset
WebSearch: "{disease}" CPTAC proteomics dataset
WebSearch: "{disease}" UK Biobank omics
WebSearch: "{disease}" AWS open data omics
Extract accession IDs, titles, and descriptions from search results. Add as rows with the
appropriate Repository label. Flag dbGaP/EGA/JGA as Access = "Controlled".
Step 13 — Classify omics type
Apply rule-based classification to each record's GEO_Type, Title, and Summary.
The GEO gdstype field is unreliable — always use title + summary as primary signal.
def classify_omics(row):
combined = (str(row.get("GEO_Type","")) + " " +
str(row.get("Title","")) + " " +
str(row.get("Summary",""))).lower()
# Single-cell (check before bulk RNA-seq)
if any(x in combined for x in ["single cell", "scrna", "10x chromium", "dropseq",
"smart-seq", "single-nucleus", "snrna"]):
return "scRNA-seq"
# Spatial transcriptomics
elif any(x in combined for x in ["spatial transcriptom", "visium", "slide-seq",
"merfish", "seqfish", "stereo-seq"]):
return "Spatial Transcriptomics"
# Chromatin accessibility
elif "atac" in combined:
return "ATAC-seq"
# CUT&RUN / CUT&TAG
elif any(x in combined for x in ["cut&run", "cut and run", "cutana", "cut&tag"]):
return "CUT&RUN/CUT&TAG"
# ChIP-seq
elif any(x in combined for x in ["chip-seq", "chip seq", "binding/occupancy",
"chromatin immunoprecipitation"]):
return "ChIP-seq"
# 3D genome
elif any(x in combined for x in ["hi-c", "hic", "3d genome", "chromatin conformation",
"capture-c", "4c-seq", "5c"]):
return "Hi-C / 3D Genome"
# DNA methylation
elif any(x in combined for x in ["methylat", "bisulfite", "wgbs", "rrbs",
"epic array", "850k", "450k", "dnmt"]):
return "DNA Methylation"
# miRNA / ncRNA
elif any(x in combined for x in ["mirna", "microrna", "ncrna", "lncrna",
"small rna", "pirna", "circrna"]):
return "miRNA/ncRNA"
# Ribo-seq / RIP-seq
elif any(x in combined for x in ["ribo-seq", "ribosome profiling", "rip-seq",
"clip-seq", "iclip"]):
return "Other (Ribo/RIP/CLIP-seq)"
# Bulk RNA-seq
elif any(x in combined for x in ["high throughput sequencing", "rna-seq", "rnaseq",
"mrna-seq", "total rna", "poly-a"]):
return "Bulk RNA-seq"
# Microarray
elif any(x in combined for x in ["expression profiling by array", "microarray",
"affymetrix", "illumina beadchip", "agilent"]):
return "Microarray"
# Genomics
elif any(x in combined for x in ["wgs", "whole genome sequencing", "whole-genome",
"snp array", "genotyping", "gwas", "exome"]):
return "Genomics (WGS/SNP/Exome)"
# Proteomics
elif any(x in combined for x in ["proteom", "mass spectrometry", "lc-ms", "tmtpro",
"label-free", "dia-ms", "dda-ms", "phosphoproteom"]):
return "Proteomics (MS)"
# Metabolomics / lipidomics
elif any(x in combined for x in ["metabolom", "metabolite", "nmr", "gcms", "lcms",
"lipidom", "lipidome", "untargeted ms"]):
return "Metabolomics/Lipidomics"
# Multi-omics
elif any(x in combined for x in ["multi-omics", "multiomics", "multi omics",
"integrat", "joint profil"]):
return "Multi-omics"
else:
return "Other/Mixed"
Step 14 — Relevance audit and final-inclusion decision
Classify every candidate with an evidence-backed relevance label and an explicit
include_in_final decision. Do not use a numeric score, confidence score, or rank score.
Do not silently drop candidates. You may exclude out-of-scope or false-positive rows from
final user-facing split tables, but excluded rows must remain auditable in the master catalog
or excluded-candidates CSV with label, reason, evidence, source, and matched terms.
Tune keyword lists for each disease — the examples below are for SCD.
POSITIVE_EVIDENCE_FIELDS = [
"Title",
"Summary",
"Description",
"Abstract",
"Publication",
"GEO_Type",
"Omics_Type",
"Technology",
"Organism",
"Species",
"Tissue",
"Cell_Type",
]
NEGATIVE_EVIDENCE_FIELDS = POSITIVE_EVIDENCE_FIELDS + ["Access"]
PROVENANCE_ONLY_FIELDS = {
"source_query",
"source_query_category",
"source_repository",
"source_api",
"retrieved_at",
"Query",
"Repository",
}
# Set from Q9. Defaults keep final user-facing tables strict and reproducible.
FINAL_TABLE_INCLUSION = "direct_only" # direct_only | direct_adjacent | direct_adjacent_weak
INCLUDE_ADJACENT_IN_FINAL = FINAL_TABLE_INCLUSION in {"direct_adjacent", "direct_adjacent_weak"}
INCLUDE_WEAK_IN_FINAL = FINAL_TABLE_INCLUSION == "direct_adjacent_weak"
def _collect_hits(row, terms, fields):
hits = []
for field in fields:
value = str(row.get(field, "") or "")
value_lower = value.lower()
for term in terms:
if term.lower() in value_lower:
hits.append({"term": term, "field": field, "value": value})
return hits
def _include_in_final(label):
if label == "direct":
return True
if label == "adjacent":
return INCLUDE_ADJACENT_IN_FINAL
if label == "weak":
return INCLUDE_WEAK_IN_FINAL
return False
def _short_evidence(hit, max_len=220):
if not hit:
return ""
value = " ".join(hit["value"].split())
term = hit["term"]
idx = value.lower().find(term.lower())
if idx == -1:
return value[:max_len]
start = max(0, idx - 80)
end = min(len(value), idx + len(term) + 120)
prefix = "..." if start > 0 else ""
suffix = "..." if end < len(value) else ""
return prefix + value[start:end] + suffix
def audit_relevance(row, direct_terms, adjacent_terms, weak_terms, out_of_scope_terms, false_positive_terms):
"""
Return required relevance columns:
relevance_label, include_in_final, relevance_reason, relevance_evidence, evidence_source, matched_terms.
direct_terms: disease/topic terms indicating patient data, primary disease biology, or a validated model
adjacent_terms: mechanistic, pathway, gene, tissue, treatment, or model terms relevant to the topic
weak_terms: broad or tangential terms that may still be useful for user review
out_of_scope_terms: terms showing the row is outside requested omics/species/tissue/scope
false_positive_terms: terms showing the row is unrelated to the disease/topic
"""
# Do not include source_query/Query/provenance fields here. They explain retrieval,
# not biological relevance.
direct_hits = _collect_hits(row, direct_terms, POSITIVE_EVIDENCE_FIELDS)
adjacent_hits = _collect_hits(row, adjacent_terms, POSITIVE_EVIDENCE_FIELDS)
weak_hits = _collect_hits(row, weak_terms, POSITIVE_EVIDENCE_FIELDS)
out_of_scope_hits = _collect_hits(row, out_of_scope_terms, NEGATIVE_EVIDENCE_FIELDS)
false_positive_hits = _collect_hits(row, false_positive_terms, NEGATIVE_EVIDENCE_FIELDS)
if false_positive_hits and not direct_hits:
label = "false_positive"
primary_hits = false_positive_hits
reason = "Excluded from final tables: false-positive term matched and no direct disease/topic evidence was found."
elif out_of_scope_hits and not direct_hits:
label = "out_of_scope"
primary_hits = out_of_scope_hits
reason = "Excluded from final tables: row is outside the requested omics/species/tissue/scope."
elif direct_hits:
label = "direct"
primary_hits = direct_hits + adjacent_hits[:3]
reason = "Directly relevant: disease/topic term matched in dataset metadata."
elif adjacent_hits:
label = "adjacent"
primary_hits = adjacent_hits
reason = "Adjacent relevance: related mechanism, gene, tissue, model, or treatment term matched in dataset metadata, but direct disease/topic evidence was not found."
elif weak_hits:
label = "weak"
primary_hits = weak_hits
reason = "Weak relevance: only broad or tangential metadata terms matched; retain for user review in the master catalog."
else:
label = "weak"
primary_hits = []
reason = "Weak relevance: retained from repository query but no configured relevance terms matched in available metadata; do not use query provenance as evidence."
matched_terms = sorted({hit["term"] for hit in primary_hits})
evidence_fields = sorted({hit["field"] for hit in primary_hits})
if label in {"direct", "adjacent"} and any(field in PROVENANCE_ONLY_FIELDS for field in evidence_fields):
raise ValueError(
f"{label} evidence used provenance-only fields {evidence_fields}. "
"Do not use source_query, Query, repository, or API metadata as relevance evidence."
)
return {
"relevance_label": label,
"include_in_final": _include_in_final(label),
"relevance_reason": reason,
"relevance_evidence": _short_evidence(primary_hits[0]) if primary_hits else "",
"evidence_source": "; ".join(evidence_fields),
"matched_terms": "; ".join(matched_terms),
}
Relevance label definitions:
| Label | Meaning | Catalog action |
|---|---|---|
direct |
Actual disease/topic samples, patient data, primary disease biology, or validated disease model | include_in_final = true |
adjacent |
Mechanistically relevant gene/pathway/treatment/tissue/model but not clearly disease/topic samples | include_in_final = false by default; set true only if the user explicitly asks to include adjacent rows in final tables |
weak |
Broad, tangential, incomplete, or metadata-sparse candidate needing user review | include_in_final = false by default; set true only if the user explicitly asks for maximum-recall final tables |
out_of_scope |
Dataset is real but outside requested omics/species/tissue/year/access scope | include_in_final = false; keep in audit trail |
false_positive |
Likely unrelated hit based on exclusion terms and lack of direct/adjacent evidence | include_in_final = false; keep in audit trail |
Example keyword lists for SCD:
direct_terms = ["sickle cell disease", "sickle cell anemia", "sickle-cell disease",
"sickle-cell anemia", "sickle cell", "HbSS", "hemoglobin S",
"vaso-occlus", "sickling", "SCA patient", "SCD patient"]
adjacent_terms = ["HbF", "fetal hemoglobin", "BCL11A", "globin switching",
"erythroid", "CD34", "hydroxyurea", "hemoglobinopathy"]
weak_terms = ["sickle cell trait", "HbAS", "beta-thalassemia", "thalassemia",
"anemia", "erythrocyte", "hemoglobin"]
out_of_scope_terms = ["non-omics", "review article", "protocol only"] # tune per run
false_positive_terms = ["lung cancer", "gastric cancer", "colon cancer",
"BACH1 lung", "unrelated disease"] # tune per run
Manually review excluded candidates in the Markdown summary. The point of the audit is transparent triage, not silent removal.
Step 15 — Assemble one master catalog
import pandas as pd
# Combine all sources
all_dfs = [df for df in [df_geo, df_sra, df_biostudies, df_pride, df_jpost,
df_metabolomics, df_cxg, df_encode, df_atlas,
df_zenodo, df_figshare, df_controlled]
if df is not None and len(df) > 0]
df_all = pd.concat(all_dfs, ignore_index=True)
# Deduplicate by accession (cross-repository duplicates are common)
df_all = df_all.drop_duplicates(subset="Accession")
# Apply omics classification
df_all["Omics_Type"] = df_all.apply(classify_omics, axis=1)
# Apply relevance audit and expand required audit fields
audit_rows = pd.DataFrame(
[audit_relevance(row, direct_terms, adjacent_terms, weak_terms, out_of_scope_terms, false_positive_terms)
for _, row in df_all.iterrows()],
index=df_all.index,
)
df_all = pd.concat([df_all, audit_rows], axis=1)
REQUIRED_RELEVANCE_COLUMNS = [
"relevance_label",
"include_in_final",
"relevance_reason",
"relevance_evidence",
"evidence_source",
"matched_terms",
]
QUERY_MANIFEST_COLUMNS = [
"source_repository",
"source_query",
"source_query_category",
"source_api",
"retrieved_at",
"n_raw_hits",
"n_records_after_dedup",
]
def require_relevance_columns(df, table_name):
missing = [col for col in REQUIRED_RELEVANCE_COLUMNS if col not in df.columns]
if missing:
raise ValueError(
f"{table_name} is missing required relevance audit columns: {missing}. "
"User-requested columns are additive, not exclusive; do not save this table."
)
if "Relevance" in df.columns and "relevance_label" not in df.columns:
raise ValueError(
f"{table_name} renamed relevance_label to Relevance. "
"Use the exact required column names."
)
return df
def require_query_manifest_columns(df):
missing = [col for col in QUERY_MANIFEST_COLUMNS if col not in df.columns]
if missing:
raise ValueError(f"query manifest is missing required columns: {missing}")
return df
# Add access level column
CONTROLLED_REPOS = {"dbGaP", "EGA", "JGA", "UK Biobank", "FinnGen"}
df_all["Access"] = df_all["Repository"].apply(
lambda r: "Controlled" if any(c in r for c in CONTROLLED_REPOS) else "Open"
)
# Sort for readability without saving any numeric score/rank column
label_order = {"direct": 0, "adjacent": 1, "weak": 2, "out_of_scope": 3, "false_positive": 4}
df_all["_label_order"] = df_all["relevance_label"].map(label_order)
df_all = df_all.sort_values(["_label_order", "Date"], ascending=[True, False]).drop(columns=["_label_order"])
# Save one CSV by default
df_all = require_relevance_columns(df_all, "master catalog")
df_all.to_csv(f"{output_dir}/{disease_slug}_omics_datasets_MASTER.csv", index=False)
query_manifest = pd.DataFrame(query_manifest_rows, columns=QUERY_MANIFEST_COLUMNS)
query_manifest = require_query_manifest_columns(query_manifest)
query_manifest.to_csv(f"{output_dir}/{disease_slug}_query_manifest.csv", index=False)
print(f"Total datasets: {len(df_all)}")
print(f"direct: {(df_all.relevance_label=='direct').sum()}")
print(f"adjacent: {(df_all.relevance_label=='adjacent').sum()}")
print(f"weak: {(df_all.relevance_label=='weak').sum()}")
print(f"out_of_scope: {(df_all.relevance_label=='out_of_scope').sum()}")
print(f"false_positive: {(df_all.relevance_label=='false_positive').sum()}")
print(f"final table rows: {(df_all.include_in_final == True).sum()}")
print(f"adjacent retained in master only: {((df_all.relevance_label == 'adjacent') & (df_all.include_in_final == False)).sum()}")
print(f"weak retained in master only: {((df_all.relevance_label == 'weak') & (df_all.include_in_final == False)).sum()}")
print(f"excluded from final: {(df_all.include_in_final == False).sum()}")
print(f"query manifest: {output_dir}/{disease_slug}_query_manifest.csv")
Step 15b — Optional split tables must preserve relevance columns
If the user asks for separate gene-expression/proteomics tables, create them only as derived
views of the audited master catalog. Do not rebuild them from scratch and do not select only
the user's requested display columns. Always append the required relevance audit columns.
Final split tables should include only rows where include_in_final == true. By default,
this means direct rows only. adjacent and weak rows remain in the master catalog unless
Q9 explicitly requested a broader final table. Excluded rows must remain in the master catalog
or be written to a separate excluded-candidates CSV.
USER_REQUESTED_COLUMNS = [
"Accession", "Title", "Species", "Tissue", "N_Samples",
"Technology", "Publication", "PMID",
]
def make_output_table(df, requested_columns, table_name):
cols = []
for col in requested_columns:
if col in df.columns and col not in cols:
cols.append(col)
for col in REQUIRED_RELEVANCE_COLUMNS:
if col not in cols:
cols.append(col)
out = df[cols].copy()
return require_relevance_columns(out, table_name)
df_final = df_all[df_all["include_in_final"] == True].copy()
df_gene_expression = make_output_table(
df_final[
df_final["Omics_Type"].isin(["Bulk RNA-seq", "Microarray", "scRNA-seq", "Spatial Transcriptomics", "miRNA/ncRNA"])
],
USER_REQUESTED_COLUMNS,
"gene expression table",
)
df_proteomics = make_output_table(
df_final[
df_final["Omics_Type"].isin(["Proteomics (MS)"])
],
USER_REQUESTED_COLUMNS,
"proteomics table",
)
df_excluded = require_relevance_columns(
df_all[df_all["include_in_final"] == False].copy(),
"excluded candidates table",
)
df_gene_expression.to_csv(f"{output_dir}/{disease_slug}_gene_expression_datasets.csv", index=False)
df_proteomics.to_csv(f"{output_dir}/{disease_slug}_proteomics_datasets.csv", index=False)
df_excluded.to_csv(f"{output_dir}/{disease_slug}_excluded_candidates.csv", index=False)
Step 16 — Generate Markdown summary report
Write <disease>_omics_summary.md with these sections:
- Overview: total candidates found, final table rows, repositories searched, date of search, whether the user requested comprehensive inventory or representative examples, final inclusion rule, raw-read aggregation mode, and query manifest path
- By repository: table of dataset counts per repository
- By omics type: table of dataset counts per omics type, split by
relevance_labelandinclude_in_finalwhen useful - Relevance breakdown: counts per label with definitions of
direct,adjacent,weak,out_of_scope, andfalse_positive; explicitly report final direct rows, adjacent rows retained in master only, weak rows retained in master only, and excluded rows - Evidence examples: representative rows for each relevance label showing
relevance_reason,relevance_evidence,evidence_source, andmatched_terms - Top direct datasets: largest N, most recent, most cited (if available); include adjacent datasets in this section only if Q9 requested adjacent rows in final tables
- Excluded candidates: counts by exclusion label (
out_of_scope,false_positive) and 5-10 representative examples with evidence and matched terms - Query provenance: count by
source_query_categoryand repository; mention exact query categories used and link/name<disease>_query_manifest.csv - Controlled-access datasets: list with access instructions
- Coverage gaps and limitations: which repositories failed, which APIs require keys, which omics types are data-sparse for this disease
Explicitly state that the CSV is an inventory with relevance annotations, not a silently
filtered list. Explicitly state that source_query is query provenance, not relevance evidence.
If rows were excluded from final tables, state that they were excluded by include_in_final=false
and are available in the master audit catalog or excluded-candidates CSV. If adjacent or weak
rows were kept out of final tables, state that they remain available in the master catalog.
If the user requested representative examples, state that the output is not exhaustive.
When rendering Markdown tables, include the required relevance audit columns even when
the user asked for specific biomedical metadata columns. It is acceptable to make the table
wide; do not omit relevance evidence to make the table shorter.
Step 17 — Generate overview figure (if requested)
Create a 3-panel figure saved as <disease>_omics_landscape.png:
import matplotlib.pyplot as plt, matplotlib
matplotlib.rcParams['font.family'] = ['Liberation Sans', 'Arimo', 'DejaVu Sans']
matplotlib.rcParams['svg.fonttype'] = 'none'
fig, axes = plt.subplots(1, 3, figsize=(18, 6))
# Panel A: Donut chart — direct datasets by omics type
# Panel B: Horizontal bar chart — all datasets by repository
# Panel C: Stacked bar timeline — datasets per year, colored by omics type
fig.savefig(f"{output_dir}/{disease_slug}_omics_landscape.png", dpi=150, bbox_inches="tight")
After saving, run Read(file_path=..., mode="media_output_check") to verify the figure renders correctly.
Known API Limitations and Workarounds
| Repository | Issue | Workaround |
|---|---|---|
| GEO | retmax=100 per query cap |
Run 20–40 targeted queries; use [Title] field tags |
| PRIDE | Response is list or dict depending on result count | data if isinstance(data, list) else data.get("_embedded", {}).get("compactprojects", []) |
| MetaboLights | Direct search API returns 404 | Use OmicsDI aggregator instead |
| Metabolomics Workbench | REST API requires exact study title match | Use keyword search via OmicsDI; use REST for known study IDs |
| ArrayExpress/BioStudies | Frequently times out | Retry with shorter timeout; most studies mirrored in GEO |
| DisGeNET | API v7 requires paid API key; returns HTML without key | Skip; use GEO + PubMed searches instead |
| GWAS Catalog | findByDiseaseTrait returns 0 for many disease names (trait name mismatch) |
Search by EFO ontology ID; browse manually at https://www.ebi.ac.uk/gwas/ |
| CellxGene | No disease-specific collection tagging for rare diseases | Filter by disease field in Census cell metadata |
| ENCODE | Rate limit 10 req/sec | Add time.sleep(0.1) between requests |
| dbGaP / EGA / JGA | No open programmatic search API | Web search + manual curation; flag as Controlled access |
| Zenodo / Figshare / Dryad | No disease-specific API | WebSearch with site: operator |
| Synapse | Requires login for some datasets | Web search for public projects; note login requirement |
| GDC/TCGA | Only relevant for cancer diseases | Skip for non-cancer diseases |
| jPOST / iProX | API may be unstable | Fall back to ProteomeCentral search at http://central.proteomexchange.org |
Scientific Caveats
-
GEO
gdstypefield is unreliable: Often says "Other" or "Expression profiling by high throughput sequencing" for ChIP-seq, ATAC-seq, and Ribo-seq studies. Always re-classify using title + summary text. Many studies are misclassified as "Hi-C" when they are RNA-seq. -
Cross-repository duplicates are common: The same study may appear in GEO, ArrayExpress, SRA, and OmicsDI. Deduplicate by accession; note that GEO accessions (GSE) and ArrayExpress accessions (E-GEOD-) often refer to the same study.
-
Controlled-access datasets: dbGaP, EGA, and JGA studies require institutional data access agreements. Include them in the catalog but clearly label as "Controlled" access.
-
Relevance audit is keyword-based: Automated labeling produces false positives (unrelated studies mentioning disease keywords) and false negatives (relevant studies with unusual terminology). Query strings are provenance only and must not be used as biological evidence. Keep all candidates in the master audit CSV and manually review
out_of_scopeandfalse_positiverows before making exclusion decisions. -
GEO retmax cap: Each individual query is capped at 100 results. For common diseases (e.g., Alzheimer's, cancer), run many targeted queries to maximize recall.
-
PRIDE sample counts: Not always reported in the API response. Report as "N/A" when missing.
-
CellxGene disease ontology: CellxGene uses MONDO ontology terms for disease annotation. Look up the correct MONDO ID for your disease before filtering cell metadata.
-
Metabolomics is data-sparse: For most rare diseases, metabolomics datasets are few. Check Metabolomics Workbench and HMDB disease pages manually if OmicsDI returns nothing.
-
Spatial transcriptomics is emerging: Most spatial data (Visium, MERFISH) is in GEO or CellxGene. The field is growing rapidly — search with recent year filters.
-
Raw-read repositories inflate counts: SRA and ENA can expose many SRR/SRX run records for one biological study. Default to study/project-level aggregation and expand run-level rows only when explicitly requested.
Example Usage
User prompt: "Find all publicly available omics datasets for Alzheimer's disease"
Skill execution:
- Synonyms:
["AD", "Alzheimer", "LOAD", "EOAD", "dementia", "amyloid", "tau"] - Tissue terms: brain, cortex, hippocampus, CSF, blood, iPSC neurons, microglia, astrocytes
- Gene terms: APOE, APP, PSEN1, PSEN2, TREM2, BIN1, CLU, ABCA7
- Run 30+ GEO queries, ArrayExpress, PRIDE, OmicsDI, CellxGene, Expression Atlas, Zenodo, Figshare
- Classify omics types, audit relevance using metadata fields only, and keep adjacent/weak rows in the master catalog by default
- Output:
alzheimers_omics_datasets_MASTER.csv,alzheimers_query_manifest.csv,alzheimers_omics_summary.md
User prompt: "Find gene expression and proteomics datasets for sickle cell disease and summarize into separate tables with accession, title, species, tissue, samples, technology, publication, PMID"
Skill execution:
- Ask search breadth / organism / year / access / final-table inclusion / raw-read granularity questions.
- Build one audited master catalog first.
- Create optional split gene-expression and proteomics CSVs as direct-only views of the master catalog by default.
- Include the user's requested columns plus
relevance_label,include_in_final,relevance_reason,relevance_evidence,evidence_source, andmatched_termsin each split CSV and Markdown table. - Do not use
source_queryas relevance evidence; use title, summary, publication, organism, tissue, technology, or other dataset metadata. - Do not rename
relevance_labeltoRelevance; do not save the split CSVs if any required relevance audit column is missing. - Save
scd_query_manifest.csv; if adjacent/weak/false-positive/out-of-scope records are excluded from the split tables, keep them in the master catalog and summarize counts with evidence.
User prompt: "What RNA-seq data is available for BCL11A in erythroid cells?"
Skill execution:
- Topic: BCL11A erythroid
- Synonyms:
["BCL11A", "CTIP1", "fetal hemoglobin", "HbF", "globin switching"] - Tissue terms: erythroid, HUDEP, CD34, erythroblast, reticulocyte
- Run targeted GEO + ArrayExpress queries; skip PRIDE/metabolomics (RNA-seq focus)
- Output:
bcl11a_erythroid_omics_datasets_MASTER.csv
User prompt: "Survey all cancer proteomics datasets for pancreatic ductal adenocarcinoma"
Skill execution:
- Synonyms:
["PDAC", "pancreatic cancer", "pancreatic adenocarcinoma"] - Query PRIDE, jPOST, iProX, GDC/TCGA (PAAD project), CPTAC, cBioPortal
- Skip metabolomics-only repos; focus on MS proteomics
- Output:
pdac_omics_datasets_MASTER.csv
Code preview
No Python/R preview files were found.
Companion files
| Type | Path | Bytes |
|---|---|---|
| Markdown | SKILL.md | 61,519 |
| JSON | skill.meta.json | 1,191 |